
AACR Annual Meeting 2018: Using Precision Medicine to Target RET-altered Cancers
While immunotherapy may be the latest tool in the treatment armamentarium for cancer, many ongoing clinical trials are assessing precision medicine approaches that target specific genetic alterations in patients’ tumors. One such clinical trial, which evaluated a selective inhibitor of an oncogenic receptor tyrosine kinase in a proof-of-concept study, was discussed at the AACR Annual Meeting 2018.
The role of RET in cancer
Receptor tyrosine kinases are key enzymes located on cell membranes that facilitate signal transduction. Due to their role in essential cellular processes such as proliferation and differentiation, mutations to these proteins can drive a wide variety of diseases. Specifically, overexpression and activating mutations to these enzymes can result in uncontrolled growth and metastasis, facilitating the development of cancer.
Certain alterations to the receptor tyrosine kinase “rearranged during transfection” (RET) can result in an active form of the enzyme, which can drive cancer formation. These alterations include activating point mutations and RET gene rearrangements, which results in the fusion of RET to a second protein. Although RET-driven cancers are rare, research has shown that RET alterations are especially common in medullary thyroid cancer (MTC), and they also occur in papillary thyroid cancer (PTC), non-small cell lung cancer (NSCLC), and other types of cancer.
As described in a recent paper published in Cancer Discovery, treatment for patients with RET-altered cancers include multikinase inhibitors (MKIs), such as cabozantinib and vandetanib. However, these drugs are not specific to RET and inhibit other tyrosine kinases more potently, resulting in off-target toxicity which can limit their dosing. Additionally, MKIs do not inhibit the RET mutations V804L/M, which are associated with acquired resistance.
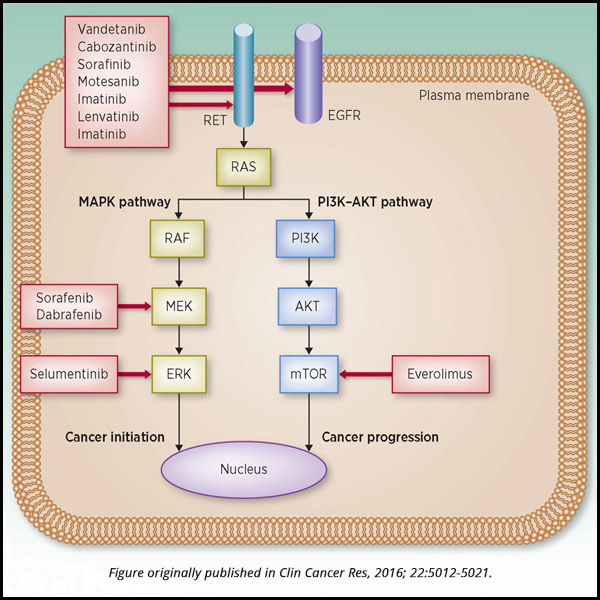
Constitutive activation or overexpression of receptor tyrosine kinases can lead to cancer. Several MKIs target tyrosine kinases, and other agents target downstream members of aberrant proliferation pathways.
BLU-667 is a selective inhibitor of RET
“RET-altered cancers across multiple tumor types represent a high medical need, as there are no approved agents that selectively target this oncogene,” said lead author of the Cancer Discovery paper, Vivek Subbiah, MD, of The University of Texas MD Anderson Cancer Center in an AACR press release. “In an effort to revolutionize treatment for these cancers, BLU-667 was designed to specifically target oncogenic RET fusions and activating mutations.”
In their study, Subbiah and colleagues generated and analyzed a library of over 10,000 kinase inhibitors and developed BLU-667, a drug that selectively targets wild-type RET and its oncogenic variants. Compared to vandetanib and cabozantinib, BLU-667 could inhibit wild-type RET with higher potency in biochemical assays. Furthermore, BLU-667 could inhibit RET V804 gatekeeper mutations with 100- to 10,000-fold higher potency compared to the other MKIs evaluated.
Many MKIs target vascular endothelial growth factor-2 (VEGFR-2), as this kinase is overexpressed in many types of cancer. However, pharmacologic inhibition of this enzyme can result in hypertension, thrombosis, and hemorrhage, the authors explain. While VEGFR-2 is not the primary target for inhibition in RET-altered cancers, Subbiah and colleagues found that vandetanib had equal potency against wild-type RET and VEGFR-2, and cabozantinib inhibited VEGFR-2 more potently than wild-type RET. Furthermore, in biochemical assays, BLU-667 was 88-fold more potent against wild-type RET compared to VEGFR-2, highlighting the selectivity of BLU-667 for RET.
The researchers also found that BLU-667 effectively suppressed signaling downstream of RET, as molecules regulated by ERK were decreased in a dose-dependent manner upon the addition of BLU-667 in cells with RET alterations.
In preclinical animal models, Subbiah and colleagues found that BLU-667 demonstrated antitumor activity in a variety of RET-altered allograft and xenograft tumors. The researchers also verified that BLU-667 did not modulate VEGFR-2 activity in vivo, further demonstrating its affinity for RET over VEGFR-2.
Evaluating BLU-667 in a first-in-human study
Subbiah and colleagues tested the selective RET inhibitor in 51 patients with RET mutations or fusions in an ongoing phase I clinical trial. These patients had MTC (29 patients), NSCLC (19 patients), PTC (two patients), or paraganglioma (a neuroendocrine cancer; one patient). Many patients had received prior therapy, including chemotherapy, immunotherapy, and MKI therapy.
As is customary in a phase I trial, patients received a range of doses of BLU-667. Evaluable patients with RET-altered cancers had an overall response rate of 45 percent, with one complete response and 17 partial responses, as presented at the AACR Annual Meeting 2018. The drug was well-tolerated, and many patients remained on treatment at the time of presentation.
One patient enrolled in this trial, Tom Cathey, showed dramatic tumor reduction after receiving BLU-667, and his story was reported by Matthew Herper in Forbes. Dose expansion for BLU-667 is currently ongoing.
“Precision targeted therapy with RET inhibition can have a powerful impact in patients whose cancer is induced by these oncogenic drivers, even in early clinical trial testing,” noted Subbiah. “I encourage all cancer patients to undergo genomic testing, as tumors with rare genomic aberrations may have effective drugs that are in clinical trials that could be beneficial to them.”

