At the AACR Annual Meeting 2026, Cancer Grand Challenges Teams Explore How Cancer Forms and Evolves
A session at the AACR Annual Meeting 2026, held April 17-22, highlighted breakthrough discoveries enabled by Cancer Grand Challenges, a global initiative cofounded by Cancer Research UK and the National Cancer Institute that supports interdisciplinary teams confronting the toughest challenges in cancer research.
The session explored “The Making of Cancer: From Cellular Events to Systemic Disease,” highlighting a research approach aimed at understanding the disease at a scale. Presenters reported on exciting findings coming out of four Cancer Grand Challenges teams that tackled the interplay of mutations, environmental cues, and epigenetics in driving tumor development and the mechanisms underlying cachexia, a syndrome that characterizes cancer as a whole-body disease.
In his introductory remarks, Paul Mischel, MD, FAACR, one of the chairs of the AACR Annual Meeting 2026 Program Committee and leader of a Cancer Grand Challenges research team, referenced the famous Francis Galton quote about nature versus nurture, drawing a parallel between the evergreen debate on the impact of these two aspects in shaping life and the role of genetics and epigenetics in cancer. “I think the work that you are going to hear about today in this session is really game changing in thinking about the interplay between the environment and our genomes and how that affects the development of cancer and its progression,” said Mischel, who is a professor at Stanford University.

In the words of session chair Charles Swanton, MD, PhD, FAACR, professor at the Francis Crick Institute in the United Kingdom, one of the beauties of Cancer Grand Challenges is that it harnesses the collective power of a global community. With presence in more than 100 institutes and more than half a billion dollars committed so far, the initiative has funded 1,800 investigators from 19 countries, taking on 18 challenges that span the breadth of the disease, noted Swanton, who is chair of the scientific committee of the Cancer Grand Challenges. He emphasized that Cancer Grand Challenges depends heavily on the generosity of philanthropic foundations, private funders, as well as national charities that have supported the initiative over the years.
“The Cancer Grand Challenges portfolio spans from understanding the earliest mutagenic changes in normal tissues, through promotion, to cancer development, to studying more advanced cancers when they have become systemic disease,” said Swanton introducing the speakers.
The Mutograph Team: Studying Biological Footprints to Identify Unknown Causes of Cancer
The first presenter was Michael R. Stratton, MBBS, PhD, FAACR, a senior group leader at Wellcome Sanger Institute in the United Kingdom, who reported on findings from the Cancer Grand Challenges project Mutographs, which he led.
The goal of Mutographs was to advance understanding of the causes of cancer through the patterns of somatic mutations that are found in normal and cancer cell genomes, said Stratton. The project was spurred by the fact that, although epidemiological evidence indicates that 80% of cancer cases are caused by lifestyle or environmental exposures, only half of them are explained by known carcinogen exposures or risk factors, and no new causes of cancer have been recently identified by conventional epidemiological approaches.
The team took an alternative strategy combining cancer epidemiology and mutation analysis to identify permanent biological “footprints” left on DNA by past carcinogen exposures and then work backwards from these footprints to identify the agents. By sequencing tens of thousands of normal and cancer genomes from 30 countries, the researchers identified a repertoire of mutational signatures, defined as patterns of DNA damage that are naturally found in human tissues.
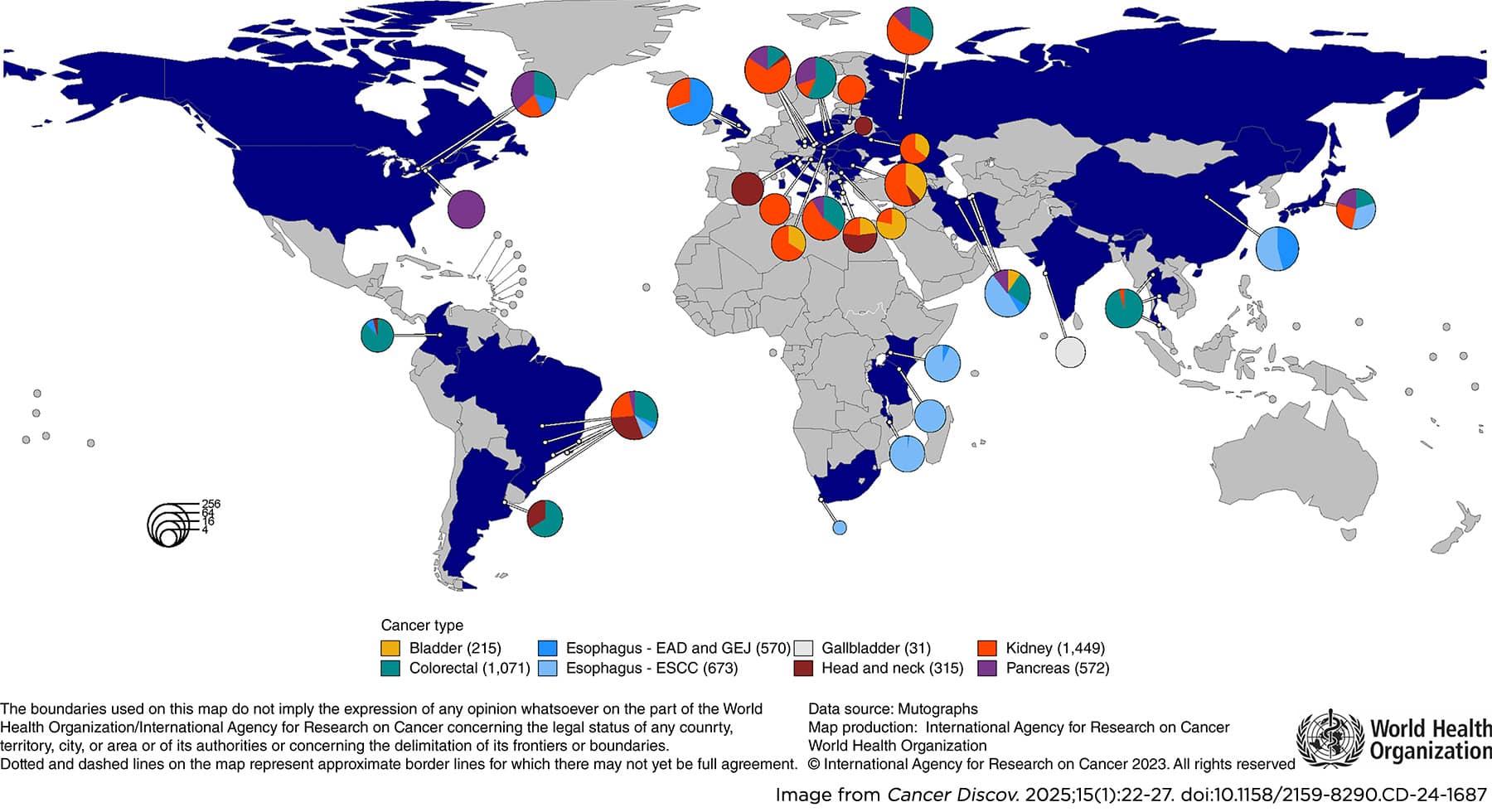
Stratton discussed three main discoveries to exemplify the many that were made by the Mutographs team over the course of eight years. Through whole-genome sequencing of hundreds of samples from patients with esophageal squamous cell carcinoma from eight countries, the researchers hoped to unravel the environmental exposure linked to the disproportionately high incidence of this type of cancer in certain parts of the world. Unexpectedly, they found that the mutational signatures of esophageal squamous cell carcinoma were very similar worldwide, suggesting that the geographical difference in incidence rates cannot be explained by exposure to a particular mutagen. This observation, Stratton pointed out, is consistent with the fact that not all carcinogens cause tumors by directly generating DNA mutations, but rather by acting as “promoters” or “selectogens” that alter the landscape in which normal cell clones carrying preexisting mutations exist and thus enabling these mutations to cause transformation.
In another study, sequencing 962 clear cell renal cell carcinomas from 11 countries with varying disease incidence, the Mutographs researchers identified associated mutational signatures, but only one of those was caused by a known plant-produced mutagen. These findings suggested the existence of multiple, geographically variable, mutagenic exposures that potentially affect tens of millions of people silently.
Lastly, Mutographs work in colorectal cancer led to the identification of geographic and age-related variations in colorectal cancer mutational processes, some of which are caused by mutagens produced by bacterial strains in the microbiome. One of these mutational signatures, associated with exposure to a mutagen called colibactin, is particularly prevalent in early-onset colorectal cancers. This led to the hypothesis that there may have been an increase in the prevalence of colibactin-producing microbes in many countries in the past 100 years and that the additional mutation burden inflicted by colibactin in the normal colorectal epithelium of children may be responsible for the increase in early onset colorectal cancer observed in the past 40 years, Stratton explained. This hypothesis is discussed in a review coauthored by Stratton and recently published in the AACR journal Cancer Discovery. The involvement of colibactin in early-onset colorectal cancer was also highlighted during the AACR Special Conference in Cancer Research: The Rise in Early-onset Cancers—Knowledge Gaps and Research Opportunities.
The PROMINENT Team: Discovering What Pushes Mutated Cells Over the Edge to Become Cancer
The next presenter was Allan Balmain, PhD, FAACR, a professor at the University of California, San Francisco, who is coleader of the Cancer Grand Challenges team PROMINENT, which seeks to understand how the environment influences cancer by causing mutated cells in normal tissues to become cancer cells. “There is a zoo of mutations and mutant clones in all of our normal tissues, and they are responding in very different ways to what we eat, what we drink, what we are exposed to,” said Balmain.
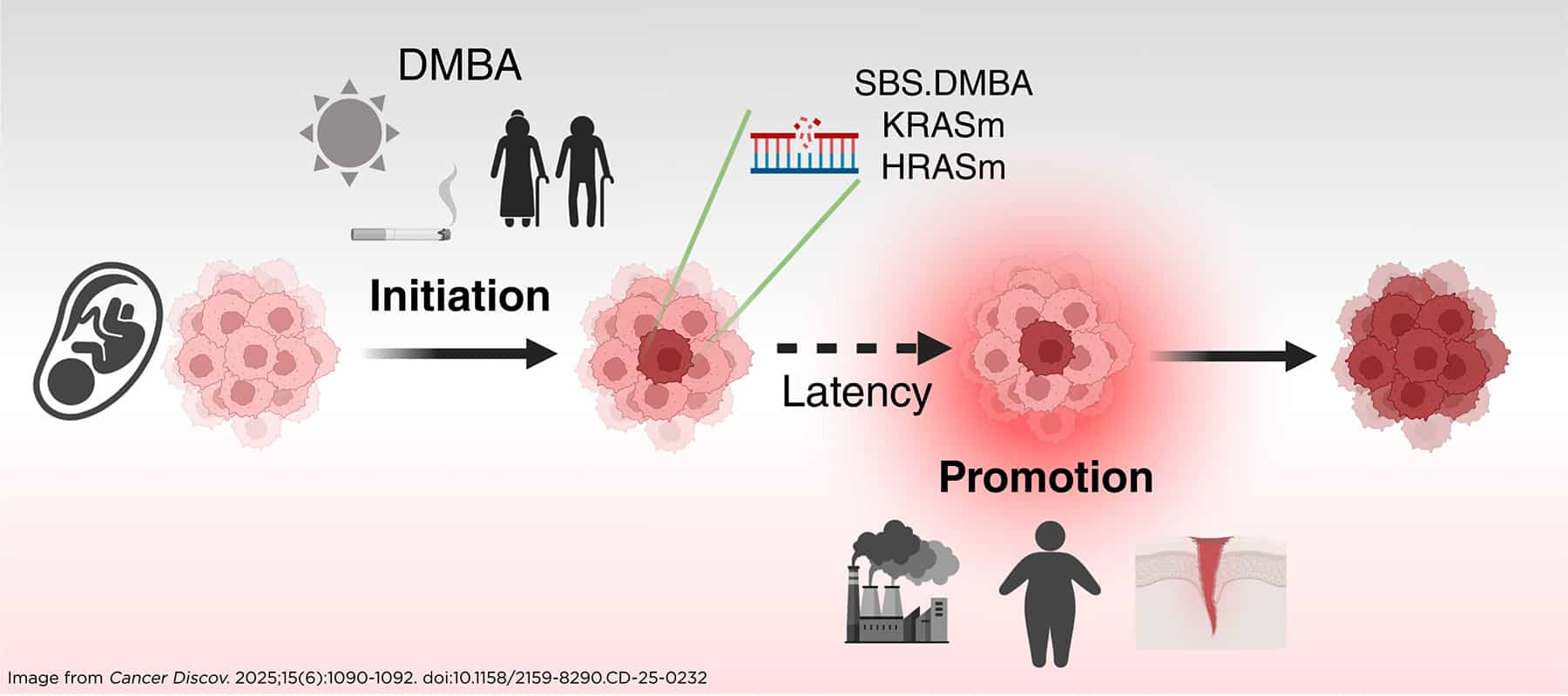
Balmain reviewed a landmark experiment conducted in the 1940s by Isaac Berenblum, MD, and colleagues that laid the foundation for the modern understanding of cancer as a multistep process. In that experiment, application of a carcinogen to the skin of mice was not enough to induce cancer; however, following that step with application of a highly inflammatory substance, which did not per se cause cancer, led to tumor development, no matter how long the interval between the two steps. This study demonstrated that substances that were later recognized as mutagens caused permanent but dormant damage that required a second hit (the “promoter” mentioned by Stratton) to cause cancer, Balmain explained.
The PROMINENT team replicated the Berenblum experiment and integrated it with whole-genome sequencing of the tumors to analyze the mutational signatures induced by the carcinogens. Through this approach, they confirmed the original observation that initiation is a necessary step for tumor development, but mutated cells can remain in the skin permanently without evidence of cancer, while the rate-limiting step in carcinogenesis is exposure to promoting factors, such as obesity, high-fat diet, and tissue wounding, said Balmain. The team recently published a review discussing this model and its implications for cancer prevention.
The SPECIFICANCER Team: Decoding the Role of Tissue Specificity
Presenter Karen M. Cichowski, PhD, a professor at Harvard Medical School, is a co-investigator on the SPECIFICANCER team that investigates why certain oncogenic mutations cause tumors only in specific tissue types. Cichowski discussed how epigenetic defects cooperate with oncogenes to drive tumor- and tissue-specific biology and how this cooperation results in therapeutic vulnerabilities that can be targeted.
“Epigenetic regulators are truly the guardians of tissue specificity, and perhaps oncogenesis in general,” said Cichowski, emphasizing that while tumors contain alterations in many oncogenic drivers, the ultimate transcriptional output of any oncogenic mutation depends on the epigenetic state of the cells, which can amplify or repress certain transcriptional responses.
Cichowski and team focused on the role of the PRC2 epigenetic complex in breast cancer, where PI3K pathway mutations are particularly frequent, especially in triple-negative breast cancer (TNBC). However, while PI3Kα and AKT inhibitors have been approved for the treatment of advanced hormone receptor-positive disease, they are not effective in TNBC. The researchers wondered whether one of the reasons TNBC tumors don’t respond to these treatments is not because the PI3K pathway is not relevant for their biology, but because the tumor cells are in a less differentiated, more basal-like state than estrogen receptor-positive luminal tumors. “Perhaps we might be able to sensitize these tumors to AKT inhibitors if we were able to somehow shift them to a more luminal state,” Cichowski asked.
The team did so by adding EZH2 inhibitors (which target the catalytic component of PRC2) to the mix. Combined treatment of different TNBC models with an EZH2 inhibitor and an AKT inhibitor caused cell death and triggered tumor regression. RNA-seq analysis showed that the two inhibitors together increased the expression of normal mature luminal markers and suppressed the expression of stem cell markers, inducing a change in cell state. This suggested that the therapy-resistant state coincided with a less differentiated state.
This observation was expanded into a broader therapeutic paradigm, said Cichowski, when the team found that EZH2 inhibitors cooperated with other targeted therapies in other tumor types, including KRAS-mutant colorectal cancer when combined with RAS inhibitors; PTEN-mutant prostate cancer when combined with AKT or mTORC1 inhibitors, and HER2-positive breast cancer when combined with HER2 kinase inhibitors. In all cases, the combined suppression of epigenetic and oncogenic pathways resulted in a shift in cell state. These findings by the SPECIFICANCER team demonstrate that certain drug combinations can kill tumors by hijacking normal tissue‑specific epigenetic pathways involved in homeostasis. These data, Cichowski emphasized, challenge the notion that drug combinations must be comprised of drugs that exhibit single-agent activity in a given tumor.
The CANCAN Team: Tackling the Whole-Body Impact of Cachexia
Eileen White, PhD, FAACR, a professor at Rutgers University and deputy director of the Rutgers Cancer Institute, concluded the session discussing work from CANCAN (Cancer Cachexia Action Network) that she coleads. Approaching cancer as a disease of the whole body, CANCAN has been working to understand the mechanisms of cachexia, a wasting syndrome that affects most patients with advanced cancer. Understanding and stopping cachexia would result in improved therapeutic outcomes but also in improved quality of life for patients, White pointed out.
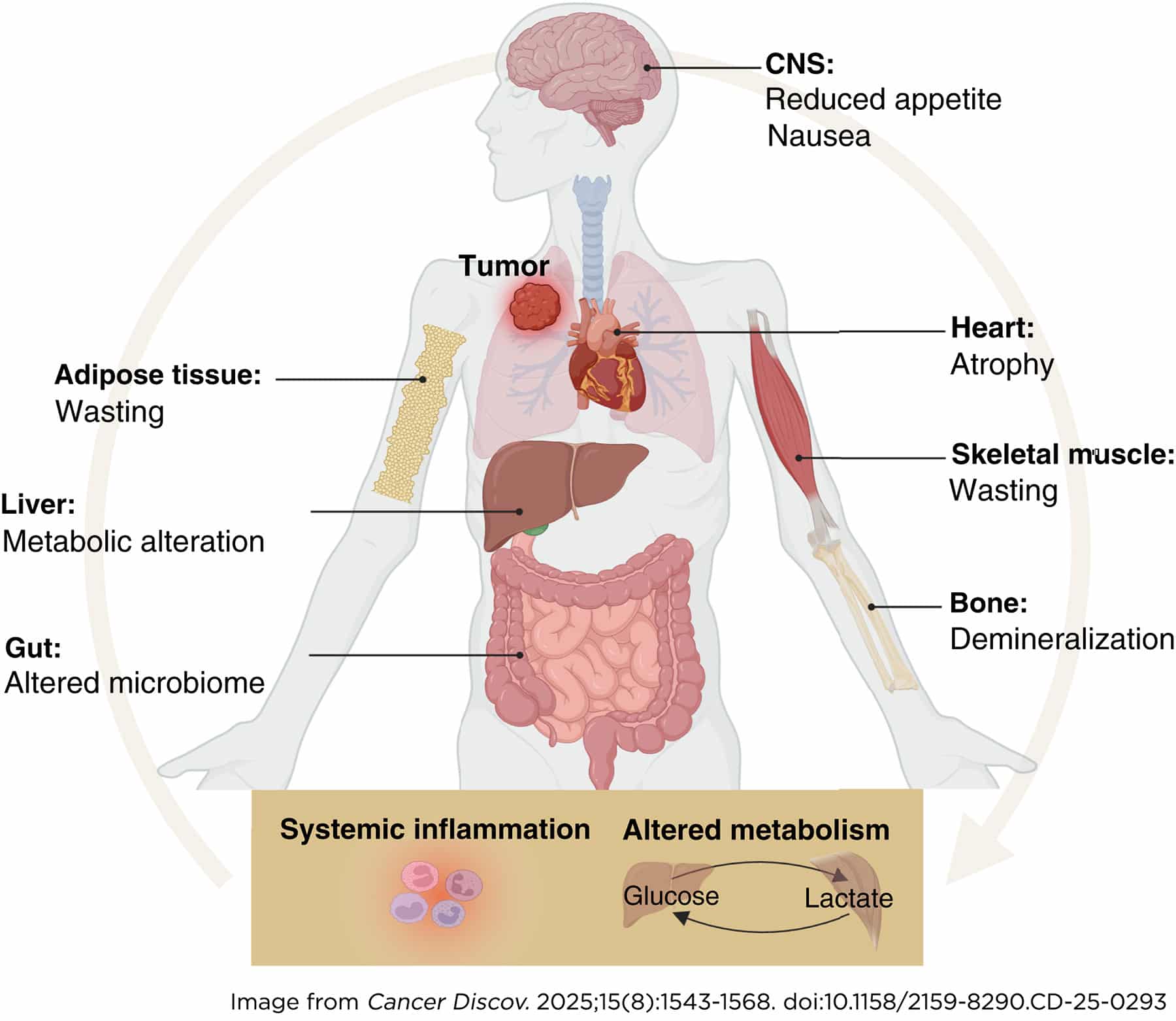
White reviewed some of the significant discoveries contributed by CANCAN member labs. Some of these discoveries relate to the identification of brain signaling drivers of cachexia. For example, a group found that the GDF15 hormone activates the endocrine stress response resulting in increased circulating levels of glucocorticoids, which can lead to muscle wasting. Another lab discovered that the cachectic factor IL6 produced by the tumor acts in the brain to limit the production of dopamine, leading to apathy, which is frequently experienced by cachectic patients along with fatigue and depression.
The White lab developed genetically engineered mouse models of cancer cachexia and utilized them to investigate metabolic alterations. Specifically, in models driven by pancreatic cancer, White and colleagues found low serotonin and high kynurenine levels, which have also been observed in cancer patients and are suggestive of a disruption in the tryptophan metabolism. White explained that tryptophan can either enter the serotonin pathway to produce serotonin or the kynurenine pathway to produce the NAD+ coenzyme, or it can be metabolized by the microbiome to produce indoles.
By favoring the kynurenine pathway, cachexia resulted in reduced production of serotonin, which may be associated with depression, lack of sleep regulation, and other symptoms experienced by cancer patients. Restoring serotonin levels in mice with a selective serotonin reuptake inhibitor (SSRI) increased food uptake and physical activity and lowered depression, suggesting that depression in cancer cachexia has a biochemical origin that may be therapeutically addressed. However, SSRI treatment had no effect on wasting, tumor burden, or survival, indicating that kynurenine induction contributes independently to cachexia.
Team CANCAN is also conducting clinical observational studies to identify and characterize different subtypes of cancer cachexia, which may help develop specific treatments. Findings from these studies pointed to the existence of two types of cachexia that manifest with different levels of stress and inflammation blood markers, different severity of symptoms, and different survival trends. Furthermore, these studies uncovered a dynamic nature of cachexia, characterized by active bouts of weight loss and periods in which weight remains stable.
AACR Annual Meeting sessions are available for virtual viewing for all registered attendees through October 2026.