Research Highlights
AACR has been the Scientific Partner of Stand Up To Cancer (SU2C) since 2008. It supports the scientific oversight, expert peer review and administration of many SU2C-funded grants. To keep the scientific community informed of the impact of this scientific partnership, a subset of research articles published by SU2C-funded scientists are summarized.
- Histone H3.3 Glycine 34-to-tryptophan (G34W) substitutions drive GCT tumorigenesis
- Nanoencapsulation enables intravenous delivery of STING agonist
- Autofluorescence imaging of macrophage dynamics in 3D culture
- T cells hampered by chemotherapy can be restored
- Glutaminase inhibitor/5-FU combination vs PIK3CA mutant CRC
- POLE mutant mice-platform for testing immune checkpoint blockade
- Targeting fusion neoantigens to treat AML
- High tumor mutation burden is associated with T cell dysfunction
- Precursor Multiple Myeloma already show signs of a compromised immune microenvironment
Histone H3.3 Glycine 34-to-tryptophan (G34W) substitutions drive GCT tumorigenesis
Title: Histone H3.3 Glycine 34-to-tryptophan (G34W) substitutions drive GCT tumorigenesis
Grant: SU2C Canada Cancer Stem Cell Dream Team
SU2C Grantee: Michael D. Taylor and Nada Jabado
Citation: Cancer Discovery, Online First version was published on November 17, 2020
Major Impact: Giant cell tumor (GCT) is a rare and aggressive bone tumor. The authors report that Glycine 34-to-tryptophan (G34W) substitutions in histone H3.3 drive GCT tumorigenesis through abnormal epigenetic remodeling. These epigenetic changes can be reversed and could be therapeutically targeted in the future.
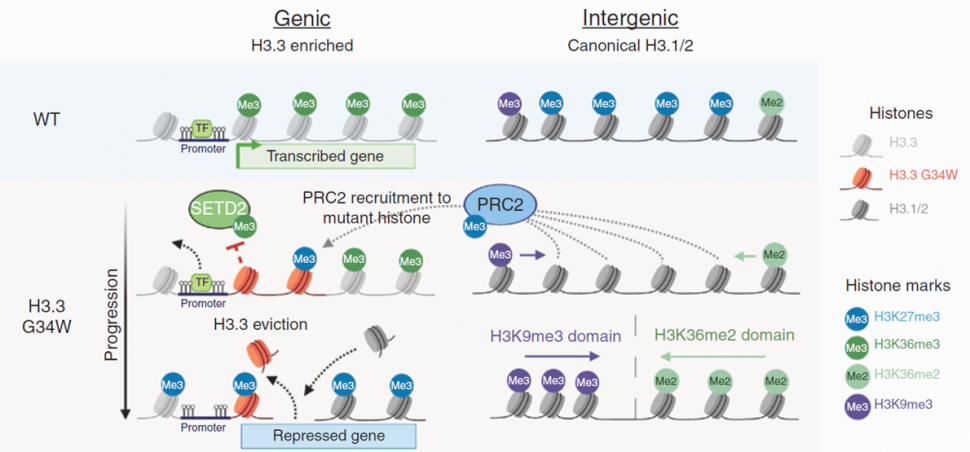
Summary: Giant cell tumor (GCT) is a rare type of bone tumor. Glycine 34-to-tryptophan (G34W) substitution in histone H3.3 is the predominant mutation occurring in GCT. To determine the role of G34W substitutions in GCT tumorigenesis, the investigators generated H3.3 G34W isogenic models using CRISPR–Cas9 technology in patient-derived stromal cells as well as patient-derived orthotopic xenograft mouse models. They found that this mutation is required for tumor formation in the GCT mouse models and also responsible for downregulation of genes involved in muscle function. The trimethylation of H3 at lysine 27 (H3K27me3) is a repressive epigenetic modification, while the trimethylation of H3 at lysine 36 (H3K36me3) is an activating chromatin mark. The investigators went on to show that the G34W substitution led to the deposition of H3K27me3 marks from intergenic to genic regions and redistribution of other chromatin marks, including H3K36me3. The investigators propose that this redistribution may create improved substrates for the repressive PRC2 complex in the genic regions, and thus promoting gene silencing. The G34W substitution was found to be essential for maintenance of proliferating osteoprogenitors and to drive the destruction of bone. The investigators are hopeful that these reversible epigenetic changes could be therapeutically targeted in the future.
Nanoencapsulation enables intravenous delivery of STING agonist
Title: Nanoparticle delivery improves the pharmacokinetic properties of cyclic dinucleotide STING agonists to open a therapeutic window for intravenous administration
Grant: Innovative Research Grant
Citation: J Controlled Release; published online November 12, 2020
Major Impact: The authors previously developed STING-activating nanoparticles (NPs) that enable the cytosolic delivery of the STING endogenous ligand, cGAMP. In this current work, they administered these STING-NPs intravenously in mouse models and confirmed that nanoencapsulation increased cGAMP’s in vivo half-life 40-fold. Significant improvements in cGAMP accumulation and STING pathway activation in the tumor, and changes in the immune cell profile of the TME were also observed. Administration of these STING-NPs significantly reduced tumor growth in melanoma and breast cancer models.

Summary: To leverage the innate immune system against cancer, efforts to engage the stimulator of interferon genes (STING) pathway have been increasingly pursued. The endogenous ligand of the STING protein, 2’3’-cyclic guanosine monophosphate-adenosine monophosphate (2’3’-cGAMP; hereafter referred as cGAMP), does not traverse the cell membrane well, and has only been efficacious when administered intratumorally. To enhance cellular uptake and cytosolic delivery, the authors previously encapsulated cGAMP in endosome-destabilizing polymer vesicles (called as STING-activating nanoparticles or STING-NPs). In this current work, they assessed the pharmacokinetics and biodistribution of intravenously administered STING-NPs. The nanoencapsulation of cGAMP increased its half-life, resulting in increased accumulation in tumor and organ (liver, kidney, spleen, and lung) tissues. These nanoparticles were more effective than “free” cGAMP in activating the STING pathway in tumors. STING-NP administration induced changes in the immune cell profile in the tumor microenvironment, including a >20-fold increase in the numbers of CD4+ and CD8+ cells. The STING-NPs significantly inhibited tumor growth in the E0771 breast cancer and B16-F10 melanoma mouse models.
Autofluorescence imaging of macrophage dynamics in 3D culture
Title: Autofluorescence imaging of 3D tumor-macrophage microscale cultures resolves spatial and temporal dynamics of macrophage metabolism
Grant: Innovative Research Grant
Citation: Cancer Research; published online October 22, 2020
Major Impact: M1 and M2 macrophages differ metabolically. M1-like macrophages, which are pro-inflammatory, upregulate glycolysis and consequently increase NADH production. On the other hand, M2-like macrophages, which are anti-inflammatory, have elevated fatty acid oxidation and oxidative phosphorylation. Furthermore, macrophage polarization is not fixed and can be influenced by the tumor microenvironment (TME). The authors developed a methodology that combines metabolic autofluorescence imaging and microscale 3D models to assess metabolic activity and visualize macrophage heterogeneity within the 3D TME. This technology can be useful in understanding tumor-associated macrophage function.

Summary: Previous studies have shown how autofluorescence imaging can be used to characterize metabolic cellular heterogeneity in vivo and in 3D cultures. Intracellular concentrations of NADH and NADPH (collectively referred to as NAD(P)H), and FAD can be determined by quantifying the endogenous fluorescence of these molecules. Furthermore, fluorescence lifetime measurements can distinguish between free and protein-bound forms of NAD(PH) and FAD. The authors interrogated the hypothesis that autofluorescence imaging of NAD(P)H and FAD can be utilized in the study of macrophage metabolism, including during polarization. By integrating 11 metabolic autofluorescence measurements from 2D cultures of macrophages stimulated with IFN- (anti-tumor M1-like phenotype) or IL-4/IL-13 (pro-tumor M2-like phenotype), the authors confirmed that macrophage polarization states can be distinguished using autofluorescence imaging. They observed metabolically distinct subpopulations (low vs high redox ratio) even within macrophages of the same polarization state. This heterogeneity was also observed in 3D macrophage monocultures and co-cultures (with tumor cells). The impact of the presence of tumors on macrophage metabolism was indicated by significant differences in NAD(P)H and FAD fluorescence lifetimes in monocultured and co-cultured macrophages. Autofluorescence imaging of macrophages as they migrated across a collagen-based extracellular matrix showed that actively migrating macrophages had lower redox ratios compared to passively-migrating macrophages, suggesting that these macrophages shifted toward oxidative metabolism as they migrated.
T cells hampered by chemotherapy can be restored
Title: Lingering effects of chemotherapy on mature T cells impair proliferation
Grant: Innovative Research Grant
Citation: Blood Advances; published online September 30, 2020

Major Impact: In pediatric leukemia, CAR T therapy’s benefits have been profound – resulting in remission rates of nearly 95%. On the other hand, as many as 25% of patients do not benefit because their T cells are not healthy enough due to the nature of their disease and/or prior chemotherapy exposure. The investigators report the deleterious effects of three chemotherapeutics on metabolic function and proliferative potential of T cells. Treating cyclophosphamide-exposed cells with N-acetylcysteine (NAC) reversed these effects. CD19 CAR T cells generated from these cyclophosphamide/NAC-treated T cells retained their function, as assessed by degranulation and IFNg production assays.
Summary: Previous observations have shown that pediatric leukemia patients have lower absolute T cell counts upon diagnosis and throughout treatment. The authors assessed the effect of three chemotherapeutic agents; namely, cyclophosphamide, cytarabine, and doxorubicin, on T-cell energy reserves, to identify a possible mechanism of poor T-cell function post-chemotherapy. Metabolic assays showed that chemotherapy significantly decreased oxygen consumption rate (OCR) and lactate conversion post-short-term T cell stimulation. To simulate the longer stimulation periods used in CAR T cell manufacturing, chemotherapy-exposed cells were allowed to recover and left unstimulated or stimulated for 7 days followed by a rest period of approximately 6 days. Under these conditions, the deleterious effects of chemotherapy on different aspects of mitochondrial respiration were evident. Given the potential importance of naïve and early memory cells on effective CAR T cell generation, the impact of chemotherapy on the different subsets was assessed. Chemotherapy almost uniformly blocked expansion of all T-cell subsets, except in the case of cyclophosphamide-treated stem central memory cells. Preliminary experiments have shown that NAC can reverse cyclophosphamide-induced cell death and mitochondrial function. Thus, CD19 CAR T cells were generated from cyclophosphamide/NAC-treated normal donor T cells and assayed for function. Although a nearly 10-fold reduction was observed in the proliferation of cyclophosphamide-exposed T cells, no differences in CAR transduction, or CAR T degranulation and IFNg production were observed.
Glutaminase inhibitor/5-FU combination vs PIK3CA mutant CRC
Title: 5-Fluorouracil enhances the anti-tumor activity of the glutaminase inhibitor CB-389 against PIK3CA-mutant colorectal cancers
Grant: Colorectal Cancer Dream Team Grant
Citation: Cancer Research; published OnlineFirst September 9, 2020

Major Impact: PIK3C mutant CRC has previously been shown to be dependent on glutamine. The authors showed that PIK3CA mutant CRC are especially susceptible to the glutaminase inhibitor CB-389. Combining 5-FU with CB-389 was found to be effective in in vivo models, paving the way for a Phase I/II clinical trial. Exploratory analysis of the Phase I data showed indications that PIK3CA mutant CRC patients may derive greater clinical benefit than patients with PIK3CA WT tumors.
Summary: The PIK3CA gene encodes the p110a catalytic subunit of PI3 kinase. Oncogenic PIK3CA mutations in colorectal cancer (CRC) cells render these cells dependent on glutamine. The authors leveraged this glutamine dependence and showed in this publication that PIK3CA mutant CRC cells are more sensitive than PIK3CA WT CRC cells to the glutaminase inhibitor CB-389. CB-389 induced upregulation of reactive oxygen species, Nrf2 nuclear translocation, and expression of uridine phosphorylase (UPP1). Given UPP1’s role in the conversion of 5-fluorouracil (5-FU) to FdUMP, the authors assessed the engagement of FdUMP with the 5-FU target thymidylate synthase using a novel cellular thermal shift assay. They found that CB-389 significantly increased FdUMP-TS binding. In vivo studies confirmed that combining CB-389 with 5-FU was more effective than single agent alone in cell line and patient-derived xenograft mouse models. In light of these promising preclinical results, the Team embarked on a phase I/phase II clinical trial in patients with different cancer types (NCT02861300). The CB-389/capecitabine (oral pro-drug form of 5-FU) combination was well tolerated at biologically active doses. Looking more closely at the tumor characteristics of the patients enrolled in the Phase I portion of the study, the Team found indications that CRC patients with PIK3CA mutations were more responsive to the drug combination when compared to other cancer types. The Team’s positive Phase I data has paved the way for the SU2C-funded Phase II portion of the clinical trial of the combination, specifically in PIK3CA mutant colorectal cancer patients.
POLE mutant mice-platform for testing immune checkpoint blockade
Title: Cancers from novel Pole mutant mouse models provide insights into polymerase-mediated hypermutagenesis and immune checkpoint blockade
Grant: SU2C-Bristol Myers Squibb Catalyst Research Grant
SU2C Grantee: Uri Y. Tabori, MD
Citation: Cancer Research; published OnlineFirst September 16, 2020
Major Impact: Animal models which can reliably mimic human mutant POLE-driven cancers are lacking and are urgently needed to study the response of polymerase mutant hypermutant cancers to immunotherapy. The Team has developed a novel mouse model which develops hypermutant cancers closely mimicking those in humans and thus provides a platform for preclinical testing of immunotherapeutics.
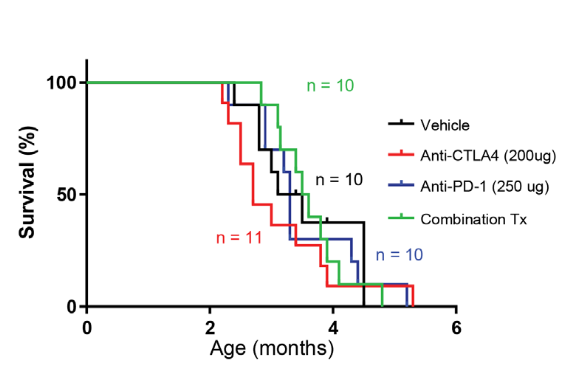
Summary: DNA polymerase epsilon (POLE) is an important member of the DNA polymerase family of enzymes. Certain POLE gene mutations have been found to be associated with hypermutant cancers. Two common mutations in the POLE gene (P286R and S459F) have been identified in several human cancers and the Team created mouse models harboring either the P286R or S459F mutation. They used these unique mouse models to ask new questions about the mechanisms of genesis in POLE mutant tumors and the correlation between genotype and cancer phenotype. They found that these POLE mutant mice mainly developed lymphomas that closely mimicked human tumors. Hypermutant cancers are usually responsive to immune check point inhibitors, but it is not clear if POLE mutant tumors would be responsive to these therapies. When the investigators treated mutant POLE-driven lymphomas in mice with immune checkpoint inhibitors (anti-PD-1 alone or in combination with anti-CTLA4), they found that these therapies did not extend their lifespan, in agreement with the human data where anti-PD1 has not shown benefit in T-cell lymphomas. Thus, these mouse models provide an opportunity to study hypermutant lymphomas and a platform to test additional immunotherapeutics.
Targeting fusion neoantigens to treat AML
Title: CBFB-MYH11 fusion neoantigen enables T-cell recognition and killing of acute myeloid leukemia
Grant: SU2C Innovative Research Grant
SU2C Grantee: Marie Bleakley, MD, PhD
Citation: J Clin Invest 2020
Major Impact: The authors provided proof of principle for the promise of targeting neoantigens even in low mutational-frequency cancers such as fusion-driven AML. They identified a CD8+ T cell epitope spanning the CBFB-MYH11 type A fusion protein and confirmed its immunogenicity. They confirmed that epitope-specific T cells were efficacious in primary AML in vitro and in vivo.
Summary: Core Binding Factor (CBF) AML arises from leukemia-initiating gene fusions, CBFB-MYH11 and RUNX1-RUNX1T1. While CBF AML can be treated with chemotherapy, many patients relapse.2 Although neoantigen-targeted therapies have been actively developed for solid tumors, such approaches have not been thoroughly leveraged against AML. Little is known about AML neoantigens and, in particular, fusion neoantigens. The researchers identified an immunogenic CBFB-MYH11 epitope that is presented on HLA-B*40:01, confirming immunogenicity using CD8+ T cells from HLA-typed healthy donors. They cloned the CBFB-MYH11-reactive T cells and confirmed that these T cells could kill primary AML cells in vitro and in vivo. As a path to developing adoptive cell therapy strategies, they sequenced the a and β chains of the TCR’s of these the CBFB-MYH11-reactive T cells, then cloned five TCRs into lentiviral vectors and transduced healthy donor CD8+ T cells. They confirmed that the TCR transfer conferred epitope specificity and anti-leukemic cytotoxicity.
High tumor mutation burden is associated with T cell dysfunction
Title: The T cell differentiation landscape is shared by tumour mutations in lung cancer
Grant: SU2C-American Lung Association-LUNGevity Lung Cancer Interception Dream Team
SU2C Grantee: Charles Swanton, MBBS
Citation: Nature Cancer 1: 546-561
Major impact: Lung cancer tissues with a higher tumor mutation burden (TMB) have a preponderance of dysfunctional T cells. This skewing towards T cell dysfunctionality may be associated with poorer survival outcomes. Characterization of these dysfunctional and terminally differentiated T cells may be used to develop new therapeutic strategies to mobilize the immune system against cancer.
Dr. James Reading, who led the study between the laboratories of Prof. Sergio Quezada and Prof. Charles Swanton explains:
“We charted the rise and fall of the immune response to lung cancer. Our bodies naturally recognise tumours as being foreign due to mutations such as those caused by smoking. When the immune system targets these mutations, it wages a war against cancer, but over time our killer cells become exhausted and their resources drained. By understanding how this process of mutation-driven immune exhaustion takes place, our work could lead to new ways of resurrecting the immune system, in particular by sustaining a fresh army of killer cells to strike at these mutations, which we believe are the Achilles heel of cancer.”

Summary: A high TMB has been associated with an increased likelihood of response to immune checkpoint inhibitors. On the other hand, antigens’ continuous presence may lead to constant or repetitive[MN1] T cell stimulation and eventual dysregulated T cell function. To explore this possibility in early stage lung cancer, the authors analyzed tumor samples from untreated patients with Stage I-IIIA disease (as part of the TRACERx study). They found that tumor tissue, especially regions with higher TMB, contained a higher percentage of dysfunctional (Tdys) and terminally differentiated T cells (TDT) than non-cancerous tissue. Consistent with dysfunction, Tdys cells expressed the immune checkpoint proteins ICOS and CTLA-4, while TDT cells were characterized by expression of Eomes and low levels of IL-7 receptor. Single-cell RNA sequencing data analyses showed that the Tdys and TDT subsets had significant transcriptional similarity with dysfunctional T cells that are observed after persistent antigen exposure. Flow cytometry and effector gene analyses suggested that these cells remained functional by virtue of granzyme B, IFNG, CD40LG expression (in CD4 Tdys cells) and TNFRSF9 expression (in CD8 Tdys cells). The abundance of dysfunctional T cells appeared to be associated with a genetic signature termed TL-DS that resulted from the loss of two proteins, TCF7 and LEF, and from differentiation skewing. Analyses of an independent set of 900 lung cancer samples indicated that the TL-DS genetic pattern was associated with higher TMB and poorer outcomes. In addition, the TL-DS gene signature was associated with survival in nine other cancer types. The findings in this study suggest that when early differentiated T cells encounter antigens in early stage cancers, a higher number of mutations may cause them to morph into dysfunctional states. Since these antigen-exposed T cells express high levels of checkpoint proteins, they may be susceptible to checkpoint inhibitors.
Learn More about our 2019 Research Highlights