
Molecular Targets and Cancer Therapeutics Conference Moves Targeting KRAS to the Fore
After three decades of failed efforts to target the elusive KRAS and its family members NRAS and HRAS, proto-oncogenes altered in about a third of cancers, encouraging data emerging from clinical trials and preclinical studies suggest KRAS could finally be dethroned from the “undruggable” category.
One of the reasons KRAS had been difficult to target is its relatively smooth surface and lack of a well-defined hydrophobic pocket in which to dock an inhibitor that can prevent its cell-signaling activity. Researchers caught a break when they discovered that a specific KRAS mutation, G12C, or a change from glycine to cysteine at position 12, is targetable. Preclinical studies showed that inhibitors that bind to cysteine at position 12 of the G12C-mutant KRAS could inhibit the downstream signaling of KRAS. About 13 percent of non-small cell lung cancers (NSCLC) and about 3 percent of colorectal cancers are known to have KRAS mutations. Mutations leading to the addition of cysteine at codon 12 account for about 50 percent of KRAS-mutant NSCLC, suggesting that targeting KRAS G12C could have a significant therapeutic impact.
The first set of promising clinical data (and updated data) came from a phase I trial, in which the KRAS G12C inhibitor AMG 510 shrank lung cancer tumors harboring KRAS G12C mutations. At the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, there was no dearth of presentations showcasing clinical and preclinical activity of an array of other KRAS inhibitors used alone or in combination with other therapeutics.
The KRAS G12C inhibitor MRTX849 shows clinical response in lung and colon cancer
Pasi A. Jänne, MD, PhD, medical oncologist at Dana-Farber Cancer Institute, presented data from a phase I clinical trial in which MRTX849, a mutant-selective small molecule KRASG12C inhibitor, was tested in patients with solid tumors.

Data from preclinical studies of this therapeutic were published simultaneously in the AACR journal Cancer Discovery. The studies showed that MRTX849 induced tumor regression in xenograft models derived from KRAS G12C-positive cell lines and patients. Combining MRTX849 with therapeutics that target receptor tyrosine kinases, mTOR, or cell cycle molecules resulted in enhanced tumor responses in many tumor models, including those resistant to MRTX849 alone. The paper also discussed data showing that MRTX849 yielded objective responses in one lung cancer and one colon adenocarcinoma patient, both of whom had KRAS G12C-positive tumors.
In a presentation prior to Jänne’s talk, James Christensen, PhD, from Mirati Therapeutics, discussed the discovery and development of MRTX849.
In an interview, Jänne noted that while MRTX849 is also a small molecule inhibitor of KRAS G12C that has activity in preclinical models and in patients with KRAS G12C mutations similar to AMG 510, there are some differences in the half-life of the two compounds. The half-life of AMG 510 is 5.5 hours, versus 25 hours for MRTX849. This may ultimately lead to clinical differences; however, the two have not been compared head to head, he cautioned.
Data from the phase I trial presented by Jänne showed that among six evaluable patients with metastatic NSCLC, three had partial responses. Of the four evaluable patients with colorectal cancer, one had a partial response. All the patients with responses received the MRTX849 dose of 600 mg twice daily.
In a poster presentation, MRTX849 was effective against a KRAS G12C tumor model as monotherapy or in combination with an anti-PD1 immune checkpoint inhibitor therapy, a type of immunotherapy now approved by the U.S. Food and Drug Administration to treat certain lung cancers. This combination led to durable, complete responses in the majority of the animals, which was not the case with monotherapy of either agents. Based on data from further studies to gain mechanistic insights, the authors hypothesize that treating the mice with the combination resulted in antitumor adaptive immune response.
Tricomplex inhibitor of the GTP-bound form of KRAS G12C offers promise
RAS proteins, including KRAS, switch between an inactive or “off” state to an active or “on” state by binding to guanosine diphosphate (GDP) and guanosine triphosphate (GTP) respectively. KRAS is predominantly GDP-bound under physiological conditions. When activated by external stimuli, the inactive RAS-GDP converts to active RAS-GTP and promotes activation of many signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway and phosphoinositide 3-kinase (PI3K) pathway.
The transition between these “off” and “on” states is regulated by guanine nucleotide exchange factors (GEFs) such as SOS1, which promote the formation of RAS-GTP, and by GTPase-activating proteins (GAPs) such as neurofibromin 1 (NF1), which promote the formation of RAS-GDP.
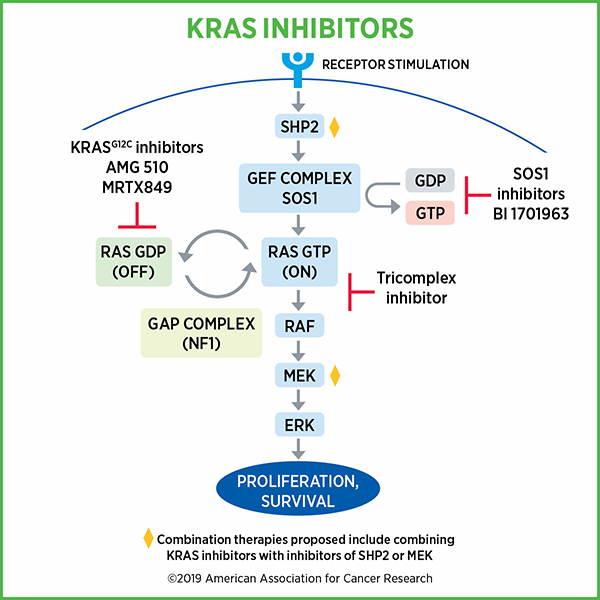
While clinical data from KRAS G12C inhibitor trials appear promising, one challenge is that these inhibitors target the inactive, GDP-bound form of KRAS G12C. Because of this, these inhibitors depend on the hydrolysis of GTP to cycle KRAS G12C proteins through the inactive, GDP-bound state for their activity. Cancer cells could potentially adapt to this situation and activate RAS by increasing upstream signaling, thereby increasing the relative abundance of GTP-bound KRAS G12C over GDP-bound KRAS G12C, and in effect, become resistant to KRAS G12C inhibitors.
In one presentation, researchers took to a unique approach to overcome the limitation of KRAS G12C inhibitors. Chris Schulze, PhD, from Revolution Medicines, discussed utilizing the immunophilin cyclophilin A as a chaperone that enables binding a KRAS inhibitor to KRAS-GTP by covalently modifying the cysteine residue of KRAS. This complex distorts the spatial arrangement of the effector face of KRAS (through steric occlusion), thus preventing it from engaging RAF, which is downstream of RAS in the process of promoting cell proliferation and growth.
Preclinical studies showed that their KRAS-GTP inhibitor lead series compound could inhibit growth of tumor xenografts, alone and in combination with an SHP2 inhibitor. SHP2 plays a role in the cycling of KRAS between “on” and “off” states. Studies also showed that inhibiting MEK increased the levels of GTP-bound KRAS through loss of negative feedback, thus, combining a MEK inhibitor with the KRAS inhibitor produced substantial tumor regression. There was also an indication that the tricomplex inhibitor approach could be effective against KRAS G13C.
SOS1 inhibitors provide another approach to inhibiting KRAS
Marco H. Hofmann, PhD, from Boehringer Ingelheim described the development of a SOS-KRAS inhibitor. Given that the binding of both mutant as well as wild-type KRAS with GTP depends on SOS1, the researchers hypothesized that selective inhibition of SOS1 will prevent the interaction of SOS1 with KRAS and inhibit KRAS irrespective of its mutational status. Hofmann showed that in preclinical studies, the SOS1-KRAS small molecule inhibitor BI-3406 specifically blocked the interaction of SOS1 with KRAS-GDP, inhibited MAPK pathway signaling, and induced tumor regression both as monotherapy as well as in combination with a MEK-inhibitor. Notably, in addition to G12C mutation, the therapeutic was effective against cell lines harboring KRAS G12D, G12V, and G13D mutations. The therapeutic was also effective against animal models of lung cancer bearing a KRAS G12S mutation and pancreatic cancer bearing a KRAS G12C mutation. Further, combining the SOS1 inhibitor with a MEK inhibitor was more effective than either therapeutic as monotherapy.
Finally, Hofmann noted that a phase I clinical trial has been initiated to test the BI-3406 analog BI 1701963 as monotherapy and in combination with the MEK inhibitor trametinib in patients with advanced solid tumors with KRAS mutation.
In another study, Benjamin Bader, PhD, from Bayer AG, discussed the development of the chemical probe BAY-293, which is a nanomolar SOS1 inhibitor that can block RAS activation. Preclinical studies showed that BAY-293 reduced the levels of GTP-bound KRAS in KRAS-wild type cells.
The authors propose combining SOS1 inhibitors with KRAS G12C inhibitors. A study published in Cancer Discovery in 2016 showed that a covalent KRAS G12C inhibitor, ARS-853, could reduce the active levels of KRAS G12C and its downstream signaling despite targeting only the inactive GDP-bound KRAS G12C. In the current presentation, they showed that BAY-293 acted synergistically with ARS-853 in inhibiting cell proliferation.
The future of KRAS inhibitors
With many more KRAS inhibitors currently being developed in addition to those described above, the field of targeting RAS has undoubtedly gone from being quiet to explosive, and this is a testament to the fast pace of research and technological advancements. However, researchers working in this space are cognizant of the likely scenario where patients develop resistance to these therapeutics. While the combinatorial treatment approaches proposed above could mitigate some of the challenges with drug resistance, it remains to be seen what impact this class of therapeutics will ultimately have on cancer patients with unmet treatment needs.
Illustration of the KRAS protein by Thomas Splettstoesser (www.scistyle.com)


