
AACR Virtual Annual Meeting I: Identifying and Targeting RAS Regulatory Mechanisms
Signaling pathways that regulate cellular proliferation are commonly mutated in cancer cells, leading to unrestrained cellular division and tumor growth. Therefore, insight into the regulation of these signaling pathways is critical to the development of novel therapeutics.
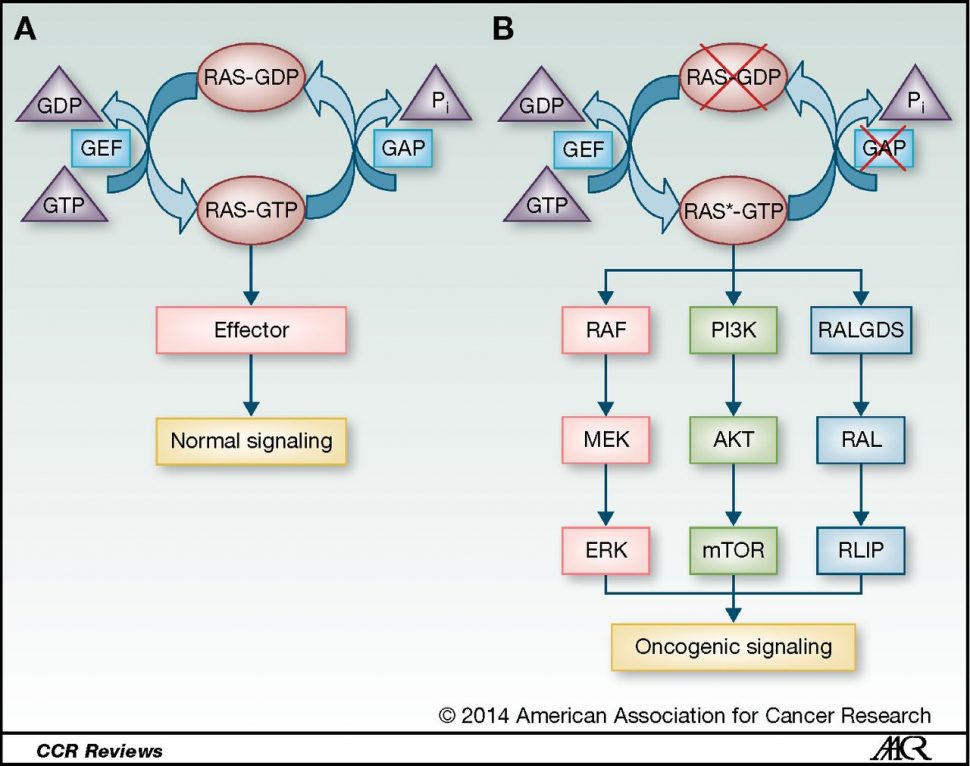
Mutations in the RAS signaling protein are found in approximately 30 percent of cancers, leading to aberrant activation of downstream signaling pathways that ultimately promote cellular proliferation. Recent advances have led to the development of multiple investigational RAS inhibitors, but to date, none have been approved for clinical use.
Studies presented at the “Emerging Signaling Vulnerabilities in Cancer” Minisymposium at the AACR Virtual Annual Meeting I uncovered novel regulatory mechanisms of RAS signaling pathways and evaluated investigational inhibitors in RAS-mutated tumor models.
Understanding regulatory mechanisms of RAS signaling pathways
EFR3A is a novel enhancer of RAS oncogenesis
A study presented by Hema Adhikari, PhD, a postdoctoral researcher at Duke University, examined RAS-interacting proteins to determine how they may impact RAS-mediated oncogenesis. Adhikari and colleagues first identified RAS-interacting proteins and found that all three RAS isoforms (KRAS, NRAS, and HRAS) interacted with ERF3A, a protein that recruits phosphatidylinositol 4-kinase alpha (PI4KA) to the plasma membrane. PI4KA generates the signaling molecule phosphatidylinositol-4-phosphate (PI4P) to activate RAS signaling.

The ERF3A gene is amplified in many RAS-mutated cancers, and an analysis of The Cancer Genome Atlas by Adhikari and colleagues revealed that high expression of ERF3A was associated with a lower probability of survival. Furthermore, they found that deletion of ERF3A or the related ERF3B in human pancreatic cancer cells decreased RAS signaling and cell viability and led to reduced tumor volume in a xenograft tumor model, suggesting that ERF3A/B promote RAS signaling and RAS-driven tumor growth.
To determine how ERF3A/B may contribute to these processes, Adhikari and colleagues examined how ERF3A/B affected an oncogenic RAS mutant. They found that ERF3A bound directly to a KRAS mutant in which the glycine at position 12 was mutated to valine (G12V), but not to wild-type KRAS or an inactive mutant. Loss of either ERF3A or ERF3B resulted in decreased levels of KRAS G12V at the plasma membrane, as well as decreased membrane levels of the signaling molecules PI4P and phosphatidylserine.
Since PI4P is generated by PI4KA, Adhikari and colleagues hypothesized that the decreased RAS signaling in ERF3A/B-deleted cells could be due to the lack of PI4KA recruitment to the plasma membrane in the absence of ERF3A/B. Consistent with this hypothesis, overexpression of a membrane-targeted PI4KA in ERF3A/B-deleted cells rescued RAS signaling and transforming ability.
Together, the results of this study suggest that the RAS-interacting proteins ERF3A/B contribute to oncogenesis by promoting membrane localization of KRAS G12V and oncogenic RAS signaling through recruitment of PI4KA to the plasma membrane.
PTEN translation is regulated by mTOR signaling
Another presentation from the session, delivered by Radha Mukherjee, PhD, a postdoctoral fellow at Memorial Sloan Kettering Cancer Center, examined regulation of the RAS- phosphatidylinositol-3-kinase (PI3K) signaling pathway. Signaling through PI3K activates AKT, which, in turn, activates mammalian target of rapamycin (mTOR), leading to cellular proliferation. AKT signaling is negatively regulated by PTEN.

Inhibitors of PI3K have had limited success, in part due to the emergence of resistance, which is associated with reactivation of AKT signaling and decreased PTEN levels, Mukherjee explained. Mukherjee and colleagues hypothesized that PTEN levels may be regulated by the PI3K pathway.
To test this hypothesis, the researchers inhibited several nodes of the PI3K pathway (HER2, PI3K, AKT, and mTOR) and examined the impact on PTEN levels. Inhibition of any of these nodes resulted in decreased PTEN levels. Further experimentation revealed that the effect of PI3K signaling on PTEN levels was due to mTOR, as inhibition of mTOR led to decreased PTEN levels independently of upstream PI3K-AKT signaling. The researchers determined that mTOR promoted translation of PTEN by phosphorylating the translation regulator 4E-BP.
These results suggest that reactivation of AKT signaling and resistance to PI3K inhibition may occur due to the reduced PTEN levels that arise when PI3K (and downstream mTOR) are inhibited. “One of the major consequences of this regulation is the reduction in efficacy of PI3K inhibitors,” said Mukherjee.
Mukherjee noted that this regulation signifies “a new feedback mechanism of the PI3K-mTOR pathway that might be an important homeostatic regulator of physiologic PI3K signaling, having major consequences for therapeutic outcomes of inhibitors of this pathway and biology of cancers driven by this pathway.”
Investigational inhibitors of SOS1
Understanding the basic regulatory mechanisms of oncogenic signaling pathways can identify potential therapeutic targets and lead to the development of novel drugs. The final study of the session, presented by Marco Hofmann, PhD, senior principal scientist at Boehringer Ingelheim RCV in Vienna, Austria, examined the impact of inhibiting an upstream regulator of RAS signaling.

Hofmann and colleagues used preclinical models to evaluate the efficacy of two investigational inhibitors of SOS1, a protein that promotes activation of KRAS. Since SOS1 is upstream of KRAS, Hofmann proposed that SOS1 inhibition could serve as a “pan-KRAS therapy.” As described in a prior blog post, Hofmann and colleagues previously reported that SOS1 inhibition led to tumor regression in preclinical models harboring various KRAS mutations and that combining SOS1 inhibition with the MEK inhibitor trametinib (Mekinist) enhanced tumor regression. A phase I clinical trial was initiated last year to evaluate the SOS1 inhibitor BI 1701963 alone and in combination with trametinib in patients with KRAS-mutated solid tumors.
The latest preclinical data reported by Hofmann showed that combining BI 1701963 with irinotecan, a DNA-damaging agent used to treat colorectal cancer, led to greater tumor regression than either inhibitor alone in KRAS-mutated models of colorectal cancer. Hofmann and colleagues found that the combination increased the levels of DNA damage and cell death markers, suggesting that SOS1 inhibition enhanced the DNA damage and cell death induced by irinotecan.
“SOS1 inhibition sensitizes KRAS-mutant tumor cells to the effect of liposomal irinotecan treatment,” said Hofmann. “These data are supportive for a combination trial.”


