
Endocrine Therapy in Breast Cancer: Making Sense of the ‘Word Salad’
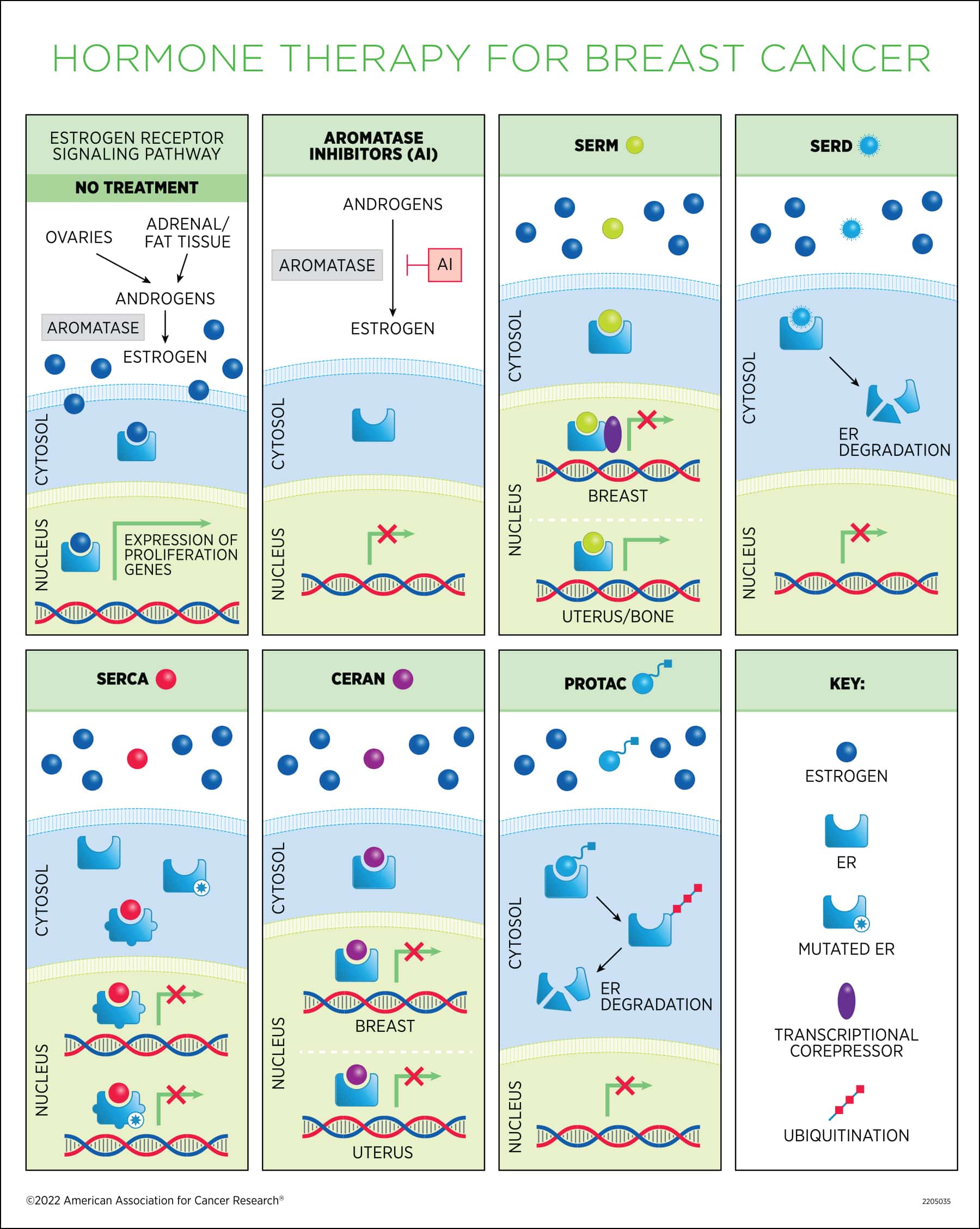
About 80 percent of breast cancers express the estrogen receptor (ER). In ER-positive tumors, estrogen is the main driver of cancer cell proliferation. Upon estrogen binding, the ER enters the nucleus and binds to DNA to activate the expression of estrogen target genes, which ultimately leads to cell cycle progression. Estrogen deprivation—blocking the body’s ability to produce estrogen or preventing estrogen from binding to the receptor—via hormone therapy is the standard of care for the treatment of ER-positive breast cancer.
In this post, we provide an overview of the different classes of hormone therapy that are used in clinical practice and the new-generation treatments that are currently under development. These were reviewed by Komal Jhaveri, MD, of Memorial Sloan Kettering Cancer Center, during a session titled “ER+ Word Salad Decoded: SERD, SERM, SERCA, CERAN, PROTAC” at the 2021 San Antonio Breast Cancer Symposium (SABCS).
The second part of this series will review data from some of the hormone therapy clinical trials that were presented at the meeting.
Approved Endocrine Therapies
Selective ER modulators (SERMs). These drugs compete with estrogen to bind to the ER and have different effects depending on the tissue. In the breast, they act as antagonists and lead to inhibition of ER-dependent transcription by recruiting transcriptional co-repressors, while in other tissues they can have agonistic activity, mimicking the effect of estrogen binding. SERMs are primarily used in premenopausal patients because estrogen levels naturally decline in menopause.
Tamoxifen (Nolvadex) was the first hormone therapy discovered. Since its FDA approval in the late 1970s, it has become the gold standard for breast cancer treatment, credited with saving countless lives around the world. Tamoxifen is commonly referred to as an antiestrogen, because it blocks the effects of estrogen in the breast tissue; however, it can also mimic the effects of estrogen in other tissues, such as uterus and bone, underlying some serious adverse events associated with long-term use.
In women who have had surgery for early and invasive breast cancer, tamoxifen lowers the chance of recurrence and improves the chances of living longer. It can be used after surgery (adjuvant therapy) or before surgery (neoadjuvant therapy) and is usually taken for at least five years. In women with metastatic breast cancer, tamoxifen can help slow cancer growth. Tamoxifen can also be used for chemoprevention in women at high risk of breast cancer. For an overview of the discovery and development of tamoxifen, see this review by V. Craig Jordan, PhD, FAACR, who discovered its breast cancer prevention properties.
Other drugs in the SERM class are toremifene (Fareston), which is structurally very similar to tamoxifen and has comparable efficacy and safety profiles, and raloxifene (Evista), a second-generation SERM that is not used for breast cancer therapy but is approved for reducing the risk of invasive breast cancer in postmenopausal women. Raloxifene can prevent bone loss due to its activity on the bone and is approved for the treatment and prevention of postmenopausal osteoporosis.
Aromatase inhibitors (AIs). First approved in the 1990s, these drugs inhibit the enzyme that catalyzes estrogen synthesis. They are used in postmenopausal patients, in whom the ovaries that are the main source of estrogen have naturally stopped producing the hormone, to block residual estrogen production in extra-ovarian tissues (fat tissue and the adrenal glands). Some premenopausal women with ER+ breast cancer can also be treated with an AI if their ovarian function is suppressed. When treating early-stage, ER+ breast cancer, AIs have less serious side effects than tamoxifen. Some women begin endocrine therapy with an AI, while others switch to one after taking tamoxifen for two to three years. Some studies are looking into whether taking an AI for five years after five years of tamoxifen could help extend the lowered risk of recurrence. Approved AIs include anastrazole (Arimidex), exemestane (Aromasin), and letrozole (Femara).
Selective ER degraders (SERDs). These compounds bind to ER, inhibit its translocation to the nucleus, and cause its destabilization and degradation. Fulvestrant (Faslodex) is the only approved SERD. Originally approved for the treatment of metastatic disease in postmenopausal patients who had developed resistance to prior endocrine therapy, its use was then extended as first-line therapy for patients with advanced or metastatic disease who have not received prior endocrine therapy. Unlike tamoxifen, fulvestrant does not display estrogen-like activity in the uterus.
Limitations of the Current Hormone Therapies
Many patients develop hormone therapy resistance over the course of treatment. Appearance of mutations in the estrogen receptor gene (ESR1) is the main mechanism of resistance and causes constitutive receptor activity in the absence of the ligand. Jhaveri explained that ESR1 mutations can be rare in patients with metastatic disease who receive hormone therapy as first-line treatment, but their prevalence is higher as detected by liquid biopsy after development of hormone therapy resistance.
In addition to ESR1 mutations, a range of other genomic alterations are also associated with resistance. To address these, endocrine agents can be combined with targeted therapies, including CDK4/6 inhibitors and PI3K inhibitors.
In addition, tamoxifen can also serve as a partial agonist and have a weaker ER suppression activity, and while fulvestrant is active after progression on prior endocrine therapy, including in some patients carrying ESR1 mutations, its efficacy is often limited by its poor bioavailability. Low therapy adherence due to side effects and other factors is also an issue. Research has shown that a considerable fraction of patients does not complete the five-year course of endocrine therapy.
The New Classes of Endocrine Therapy Agents
Research to circumvent the limitations and toxicity-related issues of the current endocrine therapy options has led to the development of new agents.
“The goal of developing all of these agents really is that we want to downregulate the ER with an agent that has the most optimal therapeutic index and superior efficacy so that we can further improve outcomes and overcome the issues we face in the clinic,” said Jhaveri.
Oral SERDs. The main advantages of this class of agents, which are the farthest along in development, are that they can be administered orally and have better adsorption and higher potency compared to fulvestrant, which can only be given as an intramuscular injection, limiting the dose that can be delivered and making the administration less practical. In addition, Jhaveri noted, oral SERDs were active in ESR1-mutant preclinical models, including those with the Y537S mutation, which was resistant to fulvestrant.
Five oral SERDs are currently being tested in phase III trials, including elacestrant, amcenestrant, giredestrant, camizestrant, and imlunestrant.
Overall, Jhaveri noted that oral SERDs are showing benefit for patients with metastatic disease, they are generally safe and well tolerated, they are effective in patients with mutant ESR1, and may become the new standard-of-care therapy post-CDK4/6 inhibitors, which represents a clinical unmet need. Oral SERDs are also being evaluated for early-stage disease.
Next-generation SERMs. Lasofoxifene is a next generation non-steroidal SERM originally developed to treat vulvovaginal atrophy and osteoporosis because of its ability to mimic estrogen in the bone. It binds to the ER with a similar potency as estrogen and causes a conformational change that prevents recruitment of cofactors. It is effective in inhibiting lung and liver metastasis in ESR1-mutant models.
Lasofoxifene is being evaluated in a phase II trial in comparison with fulvestrant for the treatment of postmenopausal and premenopausal women with locally advanced or metastatic ER-positive breast cancer and an ESR1 mutation, whose disease has progressed after treatment with the standard-of-care therapy (AI plus CDK4/6 inhibitor). Another trial is assessing the combination of lasofoxifene with the CDK4/6 inhibitor abemaciclib.
SERM/SERD hybrid. Bazedoxifene is a third-generation non-steroidal SERM that functions as an agonist in the bone (it is approved in Europe for the treatment of osteoporosis) but can also induce ER degradation in the reproductive system and is therefore considered a SERM/SERD hybrid. Bazedoxifene has shown activity in tamoxifen-resistant preclinical models and against mutant ESR1 in combination with the CDK4/6 inhibitor palbociclib. This combination is currently being evaluated in patients with ER+, endocrine-resistant metastatic breast cancer.
Selective estrogen receptor covalent antagonists (SERCAs). This class of compounds covalently inactivates both wild type and mutant ER, by targeting an amino acid residue that is conserved in both, and promotes a conformational change that represses transcription of estrogen target genes. The oral H3B-6545 is a first-in-class agent described in a Cancer Discovery article that confirmed its increased antitumor activity compared to fulvestrant in mouse models carrying both wild type and mutant ER. H3B-6545 is being investigated in phase I trials in locally advanced and metastatic breast cancer as a monotherapy and in combination with CDK4/6 inhibitors.
Complete estrogen receptor antagonists (CERANs). ER contains two domains that can activate gene transcription. Research has shown that tamoxifen is a partial ER antagonist because it only blocks one of the two activating domains. The oral small molecule OP-1250 is a first-in-class complete ER antagonist that leads to inactivation of both domains in wild type and mutant ER and has demonstrated activity in preclinical studies. OP-1250 has no agonist activity in the uterus, therefore it is not associated with increased risk of endometrial cancer. A phase I study evaluating OP-1250 in advanced breast cancer is ongoing, and early data from this trial have shown activity in ESR1 mutant patients.
Proteolysis-targeting chimeras (PROTACs). These agents exploit the body’s protein degradation machinery to degrade oncogenic proteins. They bind to the ER and recruit the E3 ubiquitin ligase, which targets the receptor for rapid and complete degradation, affording complete antagonistic activity. ARV-471 is a first-in-class PROTAC currently in phase II trial for testing as a single agent and in combination with CDK4/6 inhibition in patients with locally advanced or metastatic breast cancer.
Overall, Jhaveri concluded, these novel endocrine agents have shown activity after fulvestrant and CDK4/6 inhibitors, and on mutant ESR1. The big-picture question, she said, is figuring out a way to optimally sequence these agents to further improve outcomes for patients, both in the late- and early-stage settings, and how cost will impact their utility.
“One thing that we have learned from these agents is that, while they can address one mechanism of endocrine resistance, which is ESR1 mutation, we do require other agents and potentially other combinations to tackle the ligand binding domain mutation-independent mechanisms of endocrine resistance,” she said.
Stay tuned for part II for a deeper dive into some recent clinical trials of endocrine therapy.


