
New Molecular Targets, Technologies, and Cancer Treatments Take Center Stage at the AACR-NCI-EORTC Conference
From October 22 to 26, the Hynes Convention Center in Boston buzzed with energy as leaders in cancer drug discovery, development, and clinical trials gathered for the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. The five-day event convened more than 1,500 attendees from academia, industry, and regulatory bodies, and served as a forum for unveiling new data and exploring breakthroughs and emerging ideas in the drug development space.

Speaking about the more than 500 scientific abstracts submitted to the conference, American Association for Cancer Research (AACR) cochair of the conference, Ryan Corcoran, MD, PhD, said in his welcome address, “This reflects not just the importance of this conference, but also the great strides we are taking in innovating cancer science that will translate into novel strategies in clinical trials for the benefit of patients.” He explained that more sessions were added this year to accommodate the very high quality of abstracts received. “We developed an exciting and wide-ranging program this year to accompany these proffered abstracts to include the latest in the discovery of novel molecular targets and effective cancer therapies.”
Conference cochair Elena Garralda, MD, PhD, from the European Organisation for Research and Treatment of Cancer (EORTC), noted, “This meeting represents a unique and enduring partnership between the three institutions and it’s really an important meeting where we translate laboratory findings into clinical advances, trying to bring innovation to our patients.” The cochair from the National Cancer Institute (NCI), Tim F. Greten, MD, and several other speakers from the NCI, were unfortunately unable to participate due to the government shutdown. “But it is important to note that this program would not have been possible without Tim’s leadership and the dozens of committee members, session chairs, and speakers from the NCI,” Corcoran reiterated.
The program tackled several hot topics in the field, including targeting RAS and the latest understanding in designing and testing radiopharmaceuticals, bispecific antibodies, antibody-drug conjugates, protein degraders, cell therapies, and cancer vaccines. Presentations also revolved around emerging molecular targets and new insights into harnessing synthetic lethality, development of small molecules, and designing novel clinical trial endpoints in the era of precision medicine.
Overall, there were numerous presentations with clinical data on the newest cancer therapeutics—“an embarrassment of riches,” as Corcoran put it—many of which had innovative mechanisms of action driven by cutting-edge technologies. Here, we highlight a few presentations on investigational first-in-class therapeutics with promising early clinical trial data for patients with certain lung, prostate, colorectal, and bladder cancers. Notably, two of these drugs use novel technologies that circumvent the need for targeting driver mutations.
“It is amazing to see how the advances in technology have integrated with therapeutic development to impact cancer patients,” Corcoran said about these presentations.
HLD-0915, an orally bioavailable small-molecule RIPTAC™ for prostate cancer
Andrew Hahn, MD, from The University of Texas MD Anderson Cancer Center, presented data from a phase I/II dose escalation study testing HLD-0915, an oral small-molecule RIPTAC (Regulated Induced Proximity Targeting Chimera), for the treatment of prostate cancer.

“RIPTACs work by a ‘hold and kill’ mechanism,” explained Hahn. RIPTACs are heterobifunctional molecules that use a linker to bring a target protein selectively expressed in a cancer cell closer to an essential protein needed for normal cell functioning in order to disrupt the essential protein’s function. In HLD-0915, the target protein is the androgen receptor (AR), which is highly expressed in many cases of castrate-resistant prostate cancer, and the essential protein is BRD4, a transcription factor. The HLD-0915-mediated interaction between these two proteins leads to a stable ternary complex that prevents the transcriptional function of BRD4, thereby selectively killing the cancer cells. Because of its unique mechanism, this approach also subverts the need for the target protein to be the driver of that cancer.
Patients with metastatic, castrate-resistant prostate cancer enrolled in this trial had a median of four prior treatments, including one that targeted the AR pathway. The investigators observed antitumor activity at all doses tested, with reductions in PSA (prostate specific antigen) levels in almost 90% of the evaluable patients. All five patients with RECIST-measurable disease had partial responses, and the investigators observed reductions in circulating tumor DNA (ctDNA) fractions as well. The drug was well tolerated, and most patients continue to receive treatment, according to Hahn.
“Importantly, we see activity irrespective of whether patients had AR alterations driving their disease or if they don’t have AR alterations,” Hahn noted. “The encouraging safety and antitumor activity is proof of concept for RIPTACs in other tumor types.”
The U.S. Food and Drug Administration (FDA) issued a Fast Track designation for this therapeutic in August 2025.
FOG-001, a HELICON™ peptide that inhibits the “undruggable” WNT/β-catenin pathway
The WNT/ β-catenin cell signaling pathway has wide implications in cancer progression and prognostics, but researchers have faced numerous challenges targeting this notorious pathway. Dysregulation of this pathway causes accumulation of β-catenin in the nucleus and activation of the transcription of T-cell factor (TCF) family of transcription factors.
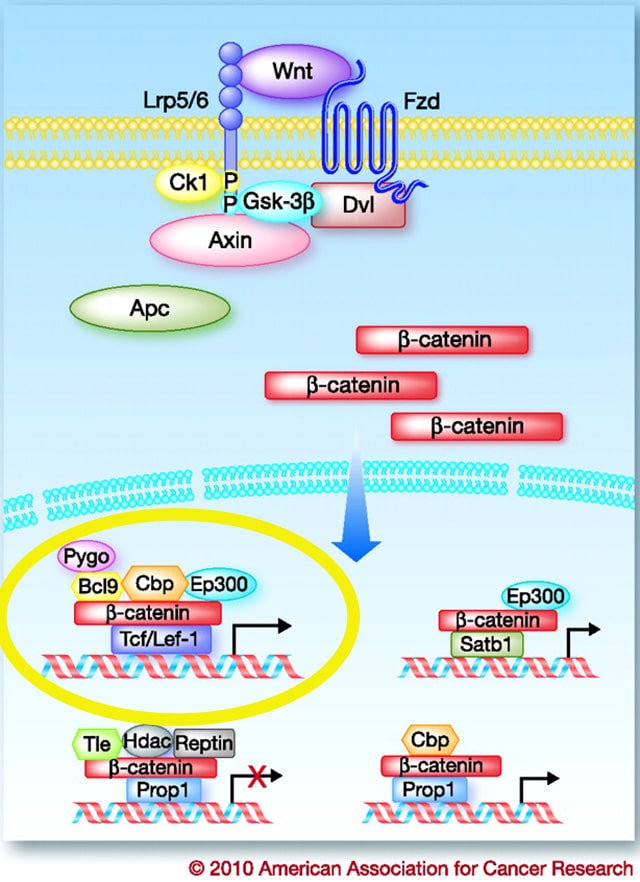
Most of the drugs developed to inhibit this pathway target the glycogen synthases (GSK-3β in the figure), but FOG-001, is designed to directly inhibit the interaction between β-catenin and the TCF family of transcription factors, with a technology called HELICON, said Samuel Klempner, MD, from Massachusetts General Hospital, who presented the study. HELICONs are α-helical peptides that can pass through cells and directly bind to β-catenin.
Here again, because this technology targets the translocation of β-catenin to the nucleus to activate TCF, the most downstream step in the WNT/ β-catenin pathway, this approach can potentially be utilized to target any mutation in the pathway regardless of whether it is a driver mutation that fuels the disease.
Klempner shared data from a phase I/II trial in which patients with microsatellite stable (MSS) colorectal cancers received this therapy. Klempner reported an overall objective response rate of 43% in patients with non-colorectal cancer and a disease control rate of 50% in efficacy-evaluable patients with MSS colorectal cancers. Biomarker analysis showed a correlation between molecular responses and increased disease stabilization rates.
“FOG-001 demonstrated a manageable safety profile with no grade 4 or grade 5 treatment-related adverse events (TRAEs),” Klempner said.
PROTAC KRAS G12D Degraders ASP3082 and ARV-806
The road to targeting RAS has been a long-winding one, but it ultimately led us to success stories in recent years when the FDA approved two therapeutics targeting KRAS G12C mutations for non-small cell lung cancers (NSCLC) and colorectal cancers. (You can read several interesting articles about this on the blog.) G12C is, however, just one of several KRAS mutations that drive different cancers. About 4% of NSCLCs and 40% of pancreatic cancers are driven by another KRAS mutation, G12D.
Currently, there are several attempts to target this mutation through the traditional route with inhibitors. A couple of presentations at the conference, however, took a different approach to tackling KRAS G12D, by degrading the protein using the proteolysis-targeting chimeras (PROTACs) technology. Like RIPTACs, these are heterobifunctional molecules that bring together, via a linker, the protein of interest, and E3 ubiquitin ligase, a protein that plays a role in a cellular process called ubiquitination that signals the degradation of the protein.

Alexander Spira, MD, PhD, from Virginia Cancer Specialists and NEXT Oncology – Virginia, shared data from a phase I trial on ASP3082, a first-in-class KRAS G12D protein degrader, specifically from the 600 mg dose cohort as that dose is being tested in phase II. Patients had previously treated, locally advanced, unresectable or metastatic, KRAS G12D-mutated NSCLC. TRAEs were seen in 96% of the patients and were mostly low grade with no drug discontinuations, Spira said. The objective response rate was 37.5% overall, with 42.9% of responses occurring in second- and third-line treatment settings. The median duration of response was 9.7 months, with about 25% of patients remaining on treatment at data cut off. Median progression-free survival was 8.25 months in second- and third-line settings.
At the 600 mg dose, there was an average of 78% degradation of KRAS G12D, Spira noted, with more than 50% reduction in KRAS G12D ctDNA levels in 63% of patients.
“ASP3082 is currently undergoing clinical evaluation as monotherapy and in combination with other cancer therapies in NSCLC, pancreatic ductal adenocarcinoma, and other cancers as well,” Spira said.
Katie Smith, PhD, from Arvinas Operations, Inc., discussed preclinical data on another PROTAC KRAS G12D degrader, ARV-806, which engages both the GTP-bound active (ON) and the GDP-bound inactive (OFF) forms of KRAS G12D. “This raises the possibility that you could essentially eliminate KRAS from the cell,” Smith said. In preclinical studies, the investigators saw subnanomolar potency across several different cell lines, including those derived from KRAS G12D-mutated NSCLC, pancreatic cancer, and colorectal cancer, and observed tumor regressions in xenograft models as well. Preclinical data also showed the drug was more potent than several KRAS G12D inhibitors currently being tested, she added.
ARV-806 is being evaluated in a phase I clinical trial in patients with KRAS G12D-mutated advanced solid tumors.
FX-909, an oral small-molecule inhibitor of PPARG for advanced urothelial carcinoma
PPAR γ (PPARG) is a transcription factor that serves as a master regulator of most urothelial cancers. High expression of PPARG is a defining feature of the luminal lineage of urothelial cancers, which represent about 65% of all patients with advanced urothelial cancers, said Xin Gao, MD, from Mass General Brigham Cancer Institute and Harvard Medical School. Researchers have long faced difficulty targeting this protein.

FX-909 is an orally bioavailable, potent small molecule that selectively inhibits both ligand-mediated and basal PPARG activity by conferring a conformationally repressive state to the transcription factor.
In a phase I clinical trial, Gao and colleagues tested FX-909 in patients with advanced solid malignancies, including locally advanced, unresectable, and metastatic urothelial carcinomas. Gao presented data from part 1a, including efficacy in patients with high expression of PPARG (PPARGhigh). The drug had an acceptable safety and tolerability profile, Gao said. Among patients with PPARGhigh, FX-909 induced tumor regression in 70% of them, which included four partial responses. Two patients experienced a complete response.
“FX-909 is a novel first-in-class drug that shows promising efficacy at all doses [in this patient population],” Gao said. “This is the first time that a targeted therapy against PPARG has shown clinical antitumor activity in urothelial cancer,” Gao noted in a press release. “We hope to further characterize the efficacy and safety profiles of FX-909 in the dose expansion study that is currently ongoing.”
Bispecifics, RNA Degraders, Antibody-drug Conjugates, and More!
Several other innovative therapeutics were also discussed at the conference, including givastomig/ABL111, a bispecific antibody that targets claudin 18.2 in the tumor and 4-1BB on adjacent T cells for certain advanced gastroesophageal cancers; REM-422, a selective, oral small-molecule degrader of MYB-encoding mRNA for recurrent or metastatic adenoid cystic carcinoma, a rare type of cancer; and ZL-1310, an antibody-drug conjugate targeting delta-like ligand-3 (DLL3) in patients with pretreated extensive-stage small cell lung cancer.
The innovative designs underpinning these therapeutics in the pipeline are just a few examples of ingenuity in the oncology drug development space that blend advanced technology and molecular insights to create the next generation of cancer treatments.


