Molecular Targets 2021: The Latest Efforts to Overcome Resistance to KRAS Inhibitors
Once considered undruggable, mutant KRAS has emerged in recent years as a viable target for anticancer therapeutics. As a member of the RAS superfamily of proteins, KRAS helps regulate cellular proliferation. When activated, KRAS upregulates proliferative pathways; when it is time to turn off cellular proliferation, KRAS is inactivated. This elegant regulatory mechanism helps prevent uncontrolled cellular proliferation and the development of cancer.
Unfortunately, this key function of KRAS can be disrupted through oncogenic mutations—most notably at codon 12—that result in the constitutive activation of KRAS, leading to uncontrolled cellular proliferation and cancer development. Such mutations are particularly common in deadly cancers such as those of the lung, colon, and pancreas, where they are mutated in 30-80 percent of cases.
Due to its prevalence and contributions to carcinogenesis, KRAS has long been considered a prime therapeutic target. However, its lack of large binding pockets hindered efforts to develop an effective inhibitor, leading critics to label KRAS as “undruggable.”
After four decades of research, the first inhibitor of KRAS, sotorasib (Lumakras) was approved for clinical use earlier this year after showing favorable results in the CodeBreaK 100 clinical trial. Sotorasib is a small molecule inhibitor that, like other investigational KRAS inhibitors, targets the inactive form of mutant KRAS, preventing it from undergoing the changes required for activation.
Notably, the May 2021 approval of sotorasib benefited from data from AACR Project GENIE, an international pan-cancer registry of real-world data. The developers of sotorasib used Project GENIE, as well as the Flatiron Health-Foundation Medicine Clinico-Genomic Database, to collect information about the demographics, response rates, and progression-free and overall survival rates of patients with KRASG12C-mutated non-small cell lung cancer (NSCLC) treated with existing therapies. Using these data, they reported to the U.S. Food and Drug Administration that patients treated with sotorasib in the CodeBreaK 100 trial had better outcomes than those historically observed in this patient population when treated with other therapies.
Based on these and other data, sotorasib received accelerated approval for the treatment of patients with KRASG12C‑mutated locally advanced or metastatic NSCLC who have received at least one prior systemic therapy. This approval provided a revolutionary new way to treat advanced NSCLC, a disease that commonly harbors KRAS mutations and is associated with poor survival rates.
Despite the marked clinical responses associated with KRAS inhibitors, the effectiveness of this therapy has been short-lived in patients due to the eventual onset of treatment resistance, which develops through several mechanisms:
Activation of upstream proteins – Since sotorasib and other KRAS inhibitors bind and inhibit only the inactive form of KRAS, one mechanism by which cancer cells overcome inhibition is by generating more of the active form of KRAS through the activation of upstream regulatory proteins. The presence of active KRAS then triggers downstream proteins and pathways that promote cellular proliferation and cancer progression.
Activation of downstream proteins and pathways – Conversely, cancer cells may bypass KRAS and directly activate downstream signaling through mutation of key signaling proteins. This allows the cell to promote proliferative signaling independently of KRAS.
Suppression of antitumor immunity – In addition to blocking proliferative signaling, inhibition of KRAS also helps stimulate components of the antitumor immune response. To overcome this effect, cancer cells may upregulate immunosuppressive proteins and processes, such as immune checkpoints, to evade antitumor immunity and potentially hinder the clinical efficacy of KRAS inhibition.
To improve the effectiveness and longevity of KRAS inhibitors, researchers are interested in combining KRAS inhibition with therapies that target these resistance mechanisms, including immunotherapies and inhibitors of upstream and downstream signaling proteins.
Researchers shared the latest clinical and preclinical results from these combinatorial approaches at the recent AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics, held October 7-10.
Targeting Proteins Upstream of KRAS to Help Overcome Resistance
Inhibition of ErbB Receptor Tyrosine Kinases
The ErbB receptor tyrosine kinases, which include EGFR, HER2, HER3, and HER4, are located at the cell membrane and are key upstream activators of KRAS. Increased stimulation of these receptor kinases—and subsequent activation of KRAS—is one mechanism by which cancer cells develop resistance to KRAS inhibitors.
Two studies presented during the meeting examined the potential of combining pan-ErbB inhibitors with KRAS inhibition in lung cancer.
In one presentation, Jacqulyne Robichaux, PhD, an assistant professor at The University of Texas MD Anderson Cancer Center, reported results from a preclinical evaluation of two inhibitors, poziotinib and afatinib (Gilotrif), that target the entire ErbB family. She demonstrated that KRAS inhibition led to the activation of all four ErbB kinases, and that the addition of either poziotinib or afatinib prevented the KRAS inhibitor-induced activation of these kinases. Furthermore, pan-ErbB inhibition synergized with KRAS inhibition to a significantly greater extent than EGFR inhibition, suggesting that inhibiting the entire ErbB kinase family may be a more effective approach than EGFR inhibition for overcoming lung cancer resistance to KRAS inhibitors.
In another presentation, David Gandara, MD, professor of medicine emeritus and director of thoracic oncology at UC Davis Comprehensive Cancer Center, reported results from the phase I CodeBreaK 101 clinical trial, which evaluated the clinical efficacy of the pan-ErbB tyrosine kinase inhibitor afatinib in combination with sotorasib in patients whose lung cancers were resistant to prior therapies. CodeBreaK 101 is also evaluating additional combination therapies with sotorasib.
The study enrolled 33 patients with advanced, KRAS-mutated NSCLC who had disease progression on prior therapies, including five patients who had progressed on prior sotorasib. Patients were divided into two cohorts, each of which received sotorasib and one of two different doses of afatinib.
Twenty percent of patients in cohort 1 and 34.8 percent of patients in cohort 2 had an objective response to the combination therapy, and at least 70 percent of patients in each cohort experienced some level of disease control. Among the five patients who had progressed on prior sotorasib treatment, three experienced stable disease with the combination therapy.
“The sotorasib/afatinib combination showed antitumor activity, including a high degree of disease control, in patients previously progressing on sotorasib alone, providing proof of principle for the rationale of combining HER family inhibitors with KRAS inhibitors,” said Gandara in an AACR press release.
Inhibition of SHP2
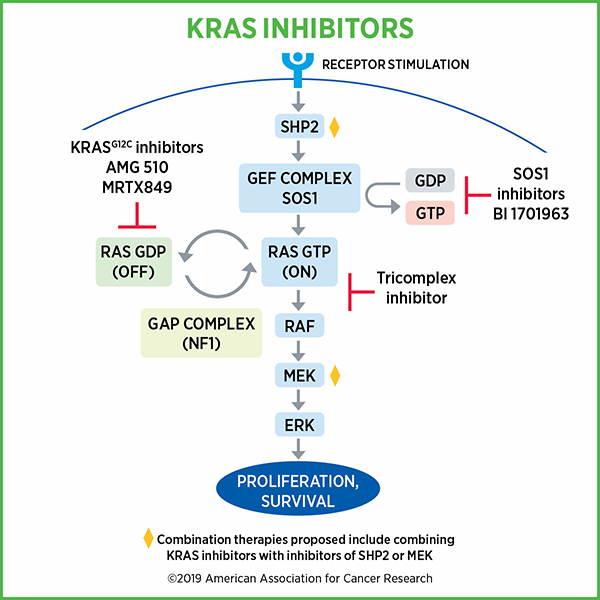
SHP2 converts the inactive form of KRAS into the activated form. Cancer cells may adapt to KRAS inhibition by increasing SHP2 activity, resulting in increased levels of activated KRAS.
In a study presented at the conference, James Stice, PhD, principal scientist at BridgeBio Pharma, tested the therapeutic potential of combining SHP2 inhibition and KRAS inhibition. Using xenograft mouse models of NSCLC, he showed that an investigational SHP2 inhibitor, BBP-398, in combination with sotorasib led to a synergistic increase in tumor regression. BBP-398 is currently under evaluation in a phase I clinical trial.
Targeting Proteins Downstream of KRAS to Help Overcome Resistance
Inhibition of Aurora Kinase A
AURKA is a protein downstream of KRAS that amplifies RAS signaling. Previous research has implicated AURKA activation in resistance to KRAS inhibition and has suggested that AURKA overexpression is associated with increased RAS signaling, greater KRAS-driven oncogenesis, and poor prognosis. In addition, inhibition of the cell cycle checkpoint protein WEE1 was previously shown to synergize with AURKA inhibition to induce cell death.
Based on these earlier observations, Jong Woo Lee, PhD, a research scientist in medical oncology at Yale Cancer Center, hypothesized that AURKA inhibition, in combination with either KRAS or WEE1 inhibition, may be an effective therapy for cancers with intrinsic or acquired resistance to the KRAS inhibitor sotorasib.
To test this hypothesis, Lee and colleagues evaluated the efficacy of the investigational AURKA inhibitor VIC-1911 in combination with sotorasib in KRAS-mutated lung cancer cells with intrinsic resistance to sotorasib; they also evaluated VIC-1911 in combination with the investigational WEE1 inhibitor adavosertib in KRAS-mutated lung cancer cells with acquired resistance to sotorasib.
Lee reported that the addition of VIC-1911 to sotorasib led to increased cell death in sotorasib-resistant cancer cells compared with the same treatment in sotorasib-sensitive cancer cells, suggesting that AURKA inhibition may help overcome sotorasib resistance. In addition, combined inhibition of AURKA and WEE1 led to a synergistic increase in the death of KRAS-mutated lung cancer cells with acquired resistance to sotorasib, as well as synergistic tumor control in KRAS/TP53-mutated lung cancer xenograft models.
“All together, these findings suggest that combination of VIC-1911 and sotorasib may represent a promising therapeutic approach for lung cancer with intrinsic resistance to direct KRASG12C inhibitors, and the combination of AURKA and WEE1 inhibition merits exploration in [cancers with] acquired resistance,” Lee concluded.
Targeting MAPK signaling
Downstream of KRAS, the MAPK signaling pathway is commonly dysregulated in cancers, leading to unrestricted cellular proliferation. Inhibition of KRAS would typically shut off MAPK signaling, but mutations in MAPK signaling proteins may allow cancer cells to bypass KRAS and activate MAPK-dependent cellular proliferation.
Suresh Ramalingam, MD, executive director of the Winship Cancer Institute at the Emory University School of Medicine, reported results from an arm of the CodeBreaK 101 clinical trial that examined whether inhibiting MEK, a MAPK signaling protein, would improve responses to KRAS inhibition.
The trial evaluated trametinib (Mekinist) in combination with sotorasib in 18 patients with KRAS-mutated colorectal cancer and 18 patients with KRAS-mutated NSCLC. The study population included patients who had received prior sotorasib therapy, as well as patients who had not.
According to Ramalingam, five of the seven patients with colorectal cancer and two of the three patients with NSCLC who had previously received sotorasib experienced stable disease when treated with the combination therapy. Among patients who had not been previously treated with sotorasib, eight of 11 patients with colorectal cancer and 10 of 15 patients with NSCLC experienced stable disease. The objective response rate for all subgroups was below 20 percent, with no complete responses observed.
“The combination of sotorasib and trametinib was safe and tolerable in patients KRASG12C-mutated solid tumors,” concluded Ramalingam. While objective response rates were lower than those previously observed with sotorasib monotherapy, Ramalingam noted that “antitumor activity was seen in patients with and without prior sotorasib therapy, and disease control was achieved by the majority of patients with prior KRASG12C inhibition.”
Considerations for Combining KRAS Inhibition with Immune Checkpoint Blockade
KRAS-mutated cancers often have several immune-suppressing mechanisms in place. Researchers are interested in understanding whether combining KRAS inhibitors with immunotherapy, such as immune checkpoint blockade, could improve the clinical benefit of KRAS inhibition.
Edurne Mugarza, PhD, a postdoctoral research assistant at the Francis Crick Institute in London, evaluated this approach in preclinical models.
Mugarza and colleagues first characterized the impact of single-agent KRAS inhibition on antitumor immunity. They showed that treatment of KRAS-mutated cancer cells with the KRAS inhibitor MRTX1257 reduced the expression of immunosuppressive cytokines and increased the expression of interferon-stimulated genes. In mice, MRTX1257 treatment led to changes in the composition of the tumor microenvironment, with a decrease in the number of immunosuppressive myeloid cells, an increase in the number of immune-activating antigen-presenting cells, and increased T-cell infiltration and activation. These results suggest that single-agent treatment with MRTX1257 may alleviate some of the immune evasion mechanisms present in KRAS-mutated tumors.
Mugarza and colleagues also observed increased expression of T-cell exhaustion markers and immune checkpoint proteins in tumors treated with MRTX1257. They, therefore, reasoned that combining MRTX1257 with immune checkpoint blockade may enhance responses in tumors with immunosuppressive tumor microenvironments.
However, combining the KRAS inhibitor with an anti-PD-1 checkpoint inhibitor did not show any therapeutic benefit over MRTX1257 alone when tested in mice with “cold” tumors, which are those with immunosuppressive microenvironments. In contrast, the combination greatly enhanced survival in mice with “hot” tumors when compared with either therapy alone.
Together, the results of this study indicate that combination treatment with MRTX1257 and immune checkpoint blockade may improve responses, but only for tumors with pre-existing antitumor immunity.
“Our study has very important clinical implications,” said Mugarza, noting that their findings in immune-resistant tumors highlight the need for alternative therapeutic options for patients with these tumors.
Looking forward
Together, the studies described here highlight the myriad of strategies being explored to improve long-term responses to KRAS inhibition. While tremendous research efforts have brought KRAS inhibitors into the clinical realm—a feat once thought impossible—there is still much to learn before the true potential of this revolutionary therapy is reached. Hopefully, continued research will soon reveal the therapeutic combinations that yield the greatest long-term benefits with minimal toxicities.
To learn about the real-life impacts of sotorasib on patient care, check out this video, which includes perspectives from a patient treated with sotorasib and from AACR President David A. Tuveson, MD, PhD, FAACR.