Hitting the cGAS: How a Cytoplasmic DNA Sensor Fights Cancer
Every cell in the human body contains about 300 million base pairs of double-stranded DNA that contribute to a range of processes throughout the cell, including gene expression, metabolic regulation, tissue identity, and protein production.
DNA’s far-reaching effects on cellular processes may make it hard to believe that it accomplishes all of its roles from a single organelle: the nucleus.
In fact, the presence of genomic DNA outside the nucleus typically indicates that something is wrong. It can mean that foreign DNA, brought in from an invading virus, is present, or that DNA damage or cellular damage has occurred.
While it has been known for some time that cytoplasmic DNA can trigger cellular defenses to thwart viral infections, namely through activation of the interferon response, the sensor of cytoplasmic DNA that triggers this response remained elusive for many years.
That was until eight years ago, when a novel sensor of cytoplasmic DNA was discovered: the cyclic GMP-AMP synthase, or cGAS.
In two back-to-back articles published in the February 2013 issue of Science, Zhijian “James” Chen, PhD, and colleagues reported the isolation of cGAS, its ability to detect cytoplasmic DNA, and the signaling cascade it activates. They found that upon recognition of cytoplasmic DNA, cGAS produces cyclic GMP-AMP (cGAMP), which then activates the stimulator of interferon genes (STING). Additional signaling promotes the expression of type I interferons, leading to the expression of various interferon-stimulated genes and subsequent activation of the immune response to fight the virus. (see Figure 1)
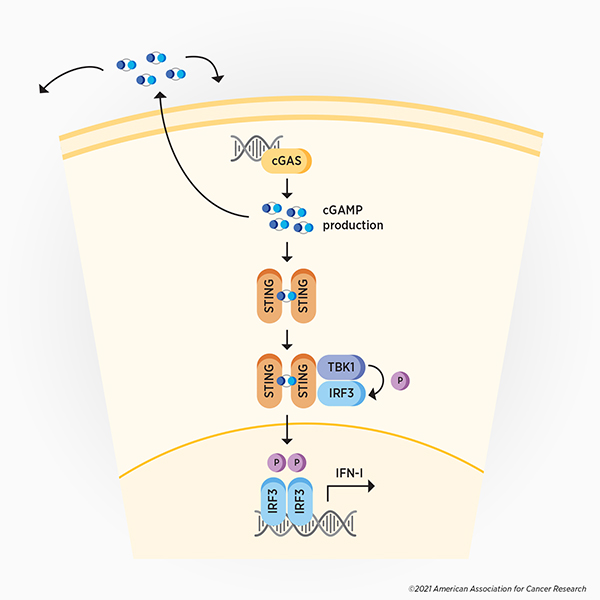
This monumental discovery uncovered a key component of the cellular defense mechanism against viruses and other cytoplasmic pathogens. Soon after, research confirming that cGAS recognizes various viruses—and that some viruses can evade or inactivate cGAS signaling—accumulated.
Since 2013, it has also become clear that the cGAS-STING pathway has roles outside the context of viral infection, contributing to cellular senescence, inflammation, and even to antitumor immunity. Similar to viruses, cancers can overcome or exploit components of this immune-activating pathway, presenting various therapeutic opportunities. Here, we review the latest research on the interplay between cancer cells and the cGAS-STING pathway.
cGAS activation promotes antitumor immunity
Recent studies have revealed how cGAS can be activated in cancer cells. Two studies published earlier this year in Cancer Cell examined the activation of antitumor immunity in cancers with mismatch repair deficiency (dMMR). One study found that defects in mismatch repair led to excess DNA digestion at DNA breaks; this resulted in chromosomal instability, export of damaged DNA into the cytoplasm, and the activation of the cGAS-STING pathway. In the accompanying study, researchers determined that such activation of cGAS in dMMR cells led to stimulation of the interferon response, promoted antitumor immunity, and enhanced the response to immune checkpoint inhibition. Consistent with these preclinical findings, the researchers observed that patients whose tumors had high levels of microsatellite instability (which is correlated with dMMR) and low levels of cGAS had worse survival. Together, the results highlight the role of the cGAS-STING pathway in stimulating antitumor immunity and suggest that the effectiveness of immune checkpoint inhibitors in dMMR tumors may be due, in part, to cGAS activation.
In addition to activating intrinsic and innate immune pathways, detection of cytoplasmic DNA by cGAS can also trigger the stimulation of adaptive immunity. In a presentation at the AACR Annual Meeting 2021, Chen, whose research group discovered cGAS in 2013, explained that cancer cells that are captured by dendritic cells release DNA and cGAMP into the cytoplasm of dendritic cells. This not only stimulates STING and downstream interferon signaling, but it also activates the dendritic cell, which then delivers tumor-associated antigens to lymph nodes, leading to the release of tumor-specific T cells.
Cancer cells fight back
The activation of the immune response by the cGAS-STING pathway can be disastrous for cancers, as components of the immune system can target and destroy malignant cells. Not surprisingly, many cancers have evolved mechanisms to overcome, evade, or exploit cGAS-mediated antitumor immunity. (see Figure 2, in red)
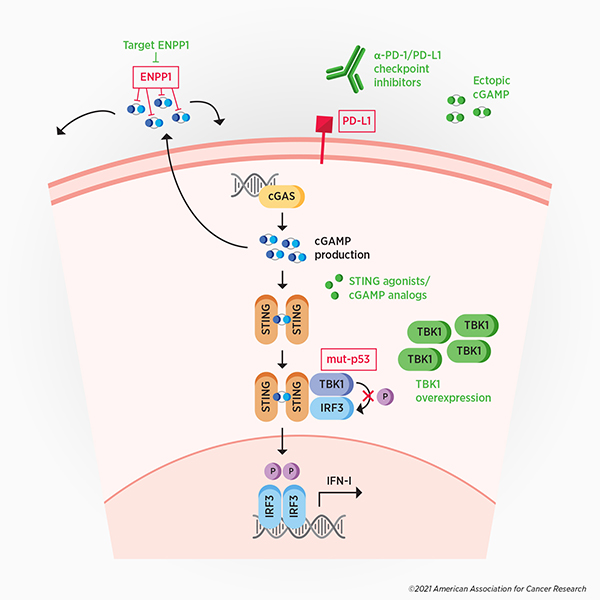
Some cancer cells evade antitumor immunity by degrading extracellular cGAMP, according to a recent study published in Cancer Discovery, a journal of the AACR. The degradation of extracellular cGAMP prevents immune signaling in neighboring cells and also generates immunosuppressive adenosine as a byproduct. This study showed that the enzyme ENPP1 ectonucleotidase is responsible for the breakdown of extracellular cGAMP, and that this enzyme is upregulated in highly metastatic cancer cells. The selective targeting of extracellular, but not intracellular, cGAMP may allow cancer cells to evade the antitumor immunity induced by extracellular cGAMP, while exploiting certain metastasis-promoting properties of intracellular cGAMP.
Another strategy by which cancer cells disrupt cGAS-mediated antitumor immunity is by suppressing the expression of interferon-stimulated genes. A study published earlier this year in Cancer Cell found that mutant p53, present in a large proportion of cancers, binds to TANK-binding kinase 1 (TBK1), a protein downstream of STING. This prevents TBK1 from phosphorylating and activating interferon regulatory factor 3 (IRF3), which, when activated, normally mediates transcription of type I interferon genes. In this way, p53-mutated cancers are able to dismantle cGAS-mediated antitumor immunity.
In addition to disrupting innate immunity, cancer cells can evade cGAS-mediated adaptive immunity by upregulating the immune checkpoint protein PD-L1 to evade activated T cells, according to remarks by Chen at the AACR Annual Meeting.
Modulating the cGAS-STING pathway for cancer treatment
The interplay between cancer cells and the cGAS-STING pathway presents various therapeutic opportunities. (see Figure 2, in green) Targeting the ENPP1 ectonucleotidase in cancer, for example, could restore paracrine immune signaling and could prevent the generation of immune-suppressing adenosine. Preclinical experiments have demonstrated that ENPP1 loss suppresses metastasis, restores immune cell infiltration in the tumor, and promotes responses to immune checkpoint inhibitors. Adenosine antagonists, which are in development, could also be used to curtail immunosuppressive strategies.
Another potential therapeutic approach is to overexpress TBK1 in p53-mutated cancers. In support of this approach, preclinical experiments found that overexpressing TBK1 was sufficient to bypass the immune-suppressing effects of mutant p53 and restore interferon-stimulated gene expression, immune cell function, and targeting of cancer cells.
In addition, components of the cGAS-STING pathway could be used as biomarkers to predict clinical responses to cancer therapies. Low cGAS levels have been shown to be associated with reduced survival of patients receiving immune checkpoint inhibitors, and ENPP1 expression was found to correlate with resistance to this immunotherapy.
Manipulation of the cGAS-STING pathway may also improve responses to existing cancer therapies. A preclinical study published in the AACR journal Blood Cancer Discovery found that STING agonists enhanced immunogenic cell death induced by bortezomib, a proteasome inhibitor used to treat multiple myeloma. While additional research is needed, the study’s findings suggest that combination treatment with bortezomib and STING agonists could benefit patients with multiple myeloma. STING activation—through either STING agonists or ectopic cGAMP treatment—has also been shown to enhance the effect of various DNA-damaging cancer therapies, including radiation, chemotherapies, and PARP inhibitors.
Data presented by Chen at this year’s AACR Annual Meeting suggest that activating the cGAS-STING pathway may also improve responses to immune checkpoint inhibition. Chen and colleagues previously observed that cGAS is required for the antitumor effect of immune checkpoint inhibition. In the research presented at the Annual Meeting, they found that an investigational cGAMP analog, which acted as a STING agonist, synergized with an anti-PD-L1 immune checkpoint inhibitor in a dose-dependent manner in mouse models of cancer. The combination treatment suppressed metastasis and sensitized anti-PD-L1-resistant cells to immune checkpoint inhibition. In addition, treatment with the cGAMP analog led to the release of chemokines associated with an immunogenic tumor microenvironment. Based on these results, a phase I clinical trial has been initiated to evaluate the investigational cGAMP analog alone or in combination with immune checkpoint inhibition in patients with various cancer types.
Fewer than 10 years have passed since the pivotal discovery of cGAS. In that short time, the known roles of this cytoplasmic DNA sensor have expanded well beyond antiviral defenses into cellular senescence and antitumor immunity, among other important functions. As we approach the end of the cGAS field’s first decade, researchers continue to study the impacts of cGAS in various cellular contexts and are exploring how to amplify the antitumor effects of the cGAS-STING pathway, with some of these exciting strategies moving into clinical trials.