
Annual Meeting 2022: KRAS, A History
When a patient receives a cancer diagnosis, doctors may perform genetic testing to determine what drives the cancer, potentially giving the patient more options for their treatment. In 2020, an estimated 13.6 percent of cancer patients in the U.S. were eligible for therapies designed to target causative mutations. This repertoire continues to grow, with 10 new targeted therapies approved in the year 2021.
This year marks the 40th anniversary of the discovery of the first human oncogenes—genes that can cause cancer when mutated to enhance their activity. This inaugural handful of genes included the infamous KRAS, which is mutated in around a quarter of human cancers but was considered “undruggable” until last year.
The journey from the discovery of KRAS to its eventual intervention was complex, requiring the efforts of countless researchers. To honor the 40th anniversary of this discovery, the AACR Annual Meeting 2022, held in New Orleans April 8-13, featured a special KRAS Anniversary Session: Novel Mechanisms for Targeting KRAS. During the session, scientists who shaped the field told the oncogene’s tale and speculated on the future of KRAS-targeted therapies.
The First Known Oncogenes
At the start of 1982, little was known about the causes of cancer aside from infection by viruses.
“Nature called 1982 the year of the oncogene, because isolation of the oncogene was not the only event—it was one of three or four major events in the identification of human transforming genes,” recalled Mariano Barbacid, PhD, FAACR, a professor of molecular oncology and former director of the Spanish National Research Center (CNIO) in Madrid. Barbacid opened the Anniversary Session by describing the history of the discovery and characterization of KRAS, in which he played a crucial role.
Researchers had known for decades that certain viruses contained genetic sequences that could stimulate dysregulated growth of host cells. In 1976, a group of scientists discovered that DNA from birds contained a gene that closely resembled the cancer-causing gene of an avian sarcoma virus. The viral gene differed in only a few places from the bird gene—mutations that may account for its oncogenic nature, the researchers argued.
Shortly thereafter, other researchers repeated this work with mammalian viruses and found that mammals also possess these “proto-oncogenic” genes. Did humans have them as well, and could they cause cancer if mutated?

Answering this question required researchers to overcome some technical challenges of the time. Namely, they wanted to prove that the transfer of DNA from cancer cells to normal cells could cause malignant changes. This technology emerged in 1979, when it was used to perform such a transfer in rodent cells, and advanced in 1981, when it was used to transfer human cancer DNA to mouse cells.
In the summer of 1982, three different labs—those of Barbacid, Robert Weinberg, PhD, FAACR, and Michael Wigler, PhD, FAACR—independently isolated and sequenced the first “transforming sequence,” a homolog of the viral Hras oncogene, from a bladder carcinoma cell line. The sequence was named HRAS as a result. A month later, the lab of Geoffrey Cooper, PhD, discovered a second transforming sequence called KRAS.
“These discoveries validated a lot of work done by molecular biologists that, at the time, was not appreciated by clinicians,” Barbacid said. “The work done with retroviruses was not just entertainment for basic scientists; it established the basis for understanding the molecular biology of human cancer.”
Once they had identified these genes, they could study their sequences to determine what made them oncogenic. Barbacid, Weinberg, and Wigler independently sequenced KRAS and made a discovery that shocked most scholars of the time, according to Barbacid: A single point mutation was sufficient to turn KRAS into a cancer driver.
The discovery was so striking that many scientists questioned it; perhaps the mutation was not a tumor driver but rather an artefact of long-term culture of the cells used in the experiment. Amidst the controversy, Barbacid and colleagues were already working to demonstrate their theory in human patients. They managed to isolate a handful of oncogenes from human tumors, which led them to the discovery of the first non-Ras oncogene, TRK. Further, by comparing the sequences of tumor-derived Ras with sequences of Ras isolated from normal tissue, the researchers found that mutant Ras was only found in malignant cells.
“This really put a nail in the coffin for those thinking the mutation had nothing to do with human cancer,” Barbacid said.
The Function of KRAS
Identifying the first human oncogenes was only the tip of the iceberg. The vast potential unlocked by these discoveries inspired younger scientists, such as Frank McCormick, PhD, FRS, FAACR, to understand how Ras proteins work in both a normal and mutated state.
McCormick, now the David A. Wood Chair of Tumor Biology and Cancer Research at the University of California San Francisco, picked up this mantle with gusto. “When I learned that a single amino acid change could convert a normal protein into an oncoprotein with such profound biological outcomes, I thought it would be extremely interesting to find out how that actually happened,” he said.

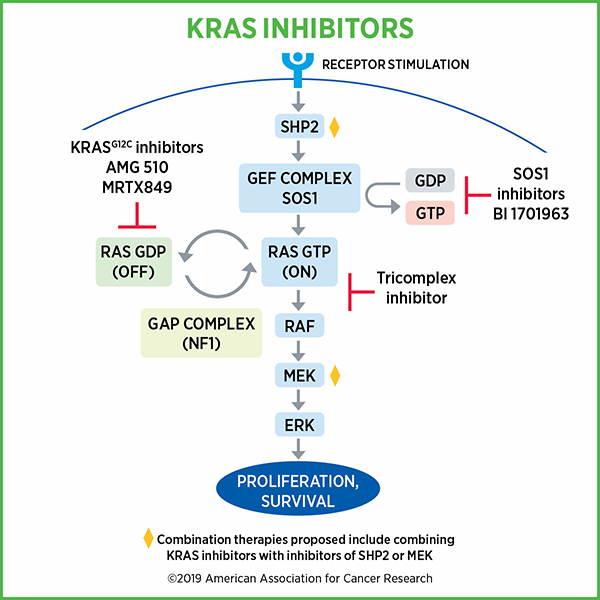
The function of normal Ras seemed straightforward, McCormick recalled. Ras proteins activate a single protein signaling pathway, the MAPK pathway. They oscillate between active and inactive forms using two simple switches, a process that McCormick helped discover. Enzymes called guanine exchange factors activate Ras by swapping out the depleted energy molecule GDP for the charged energy molecule GTP in the Ras binding pocket. Ras can then be deactivated by GTPase activating proteins, which help KRAS turn GTP into GDP.
Activated Ras proteins trigger a fairly linear signaling cascade that promotes cell growth, and their activity is tightly regulated by the enzymes that switch it on and off. Ras mutations, however, block GTPase activating proteins from reaching the binding pocket, which locks Ras in a permanently activated state.
“These proteins are generally resistant to the major ‘off’ switch and therefore get stuck in the ‘on’ state, signaling downstream consistently,” McCormick said.
McCormick also explained that mutated Ras proteins can activate protein signaling pathways outside the MAPK cascade, which adds further complexity. Nevertheless, when he and his colleagues made these discoveries in the mid-to-late 1980s, McCormick believed that drugs targeting mutant KRAS were on the horizon, rather than a full 30 years away.
“I thought naively that it would be relatively straightforward to find drugs that turned off the mutant protein but not the wild-type protein,” he said, foreshadowing the issues that would preclude such discoveries.
What, then, made KRAS so undruggable, and how did researchers overcome the challenges?
Drugging the Undruggable
A common mechanism of inhibiting oncogenic proteins is to prevent binding of the factors that activate the protein. In the case of KRAS, researchers scrambled to design drugs that would block GTP from entering the binding pocket. A series of setbacks in the early 1990s, however, demonstrated that this would be a far more laborious process than originally imagined.
Researchers found that the binding pocket of KRAS had a remarkably strong affinity for GTP, making it virtually impossible to design a drug that could outcompete it. When they searched for other pockets or grooves a drug might fit into, they found the outer surface of KRAS to be quite smooth.
“The most beautiful binding pocket on the protein is essentially off limits because of the biophysical high-affinity interaction,” said Kevan Shokat, PhD, a professor in the department of Cellular and Molecular Pharmacology at the University of California San Francisco, a professor in the Department of Chemistry at the University of California Berkeley, and a Howard Hughes Medical Institute investigator.
In 2013, Shokat and his team discovered the first compounds capable of binding to mutant KRAS by leveraging a unique property of one of the most common KRAS mutations, called G12C. The cysteine amino acid in the mutated protein forms a promising substrate for inhibitors to latch on, and because the compounds would specifically target the mutant cysteine, they would spare normal KRAS in noncancerous tissues.

“Whenever chemists see a cysteine, regardless of what the protein looks like, there’s a chemical covalent opportunity to make an irreversible bond to the target,” Shokat said. “That attracted our attention immediately.”
Shokat and colleagues assessed the binding of a library of cysteine-targeting compounds to KRAS G12C and found a few that stuck. By studying these compounds further, Shokat noticed that the mutant cysteine was located in a small binding pocket only accessible in the inactive conformation of KRAS.
He and his team then made modifications to the selected compound, giving it more flexibility to fit into the groove and making it bulkier so the protein couldn’t move around it. Their first attempts produced encouraging results in cell cultures, but they were not effective in animal models. After a few more edits to the structure, Shokat and colleagues had the backbone for sotorasib (Lumakras), the first inhibitor of mutant KRAS that would eventually be approved by the U.S. Food and Drug Administration for clinical use in 2021.
Insights for the Future
Although an approved inhibitor reflects marked progress in the field of KRAS research, the story does not end there. Challenges such as the development of treatment resistance and limited applicability to mutations beyond G12C will require additional efforts to overcome.
Each presenter provided their thoughts on the future of KRAS, supported by data from recent advances. Barbacid described a study in which his team stimulated lung tumor formation in mice, then genetically removed KRAS from the lung tissue. In every case, KRAS deletion resulted in complete tumor regression, even when they used a model that also included non-KRAS mutations. This, Barbacid said, suggests that pharmacological degradation of mutant KRAS could form a promising therapeutic strategy.
Given that mutant KRAS can activate other oncogenic pathways, such as the PI3K pathway, McCormick suggested that targeting the crosstalk between pathways could have a potential benefit. Although the PI3K inhibitor alpelisib is approved for the treatment of certain breast cancers, targeting of both KRAS and PI3K could cause significant toxicity, McCormick argued. Instead, he and his colleagues are working to develop drugs that can inhibit the interaction between KRAS and PI3K.
Shokat focused on the targeting of other common mutations at the G12 position of KRAS, such as G12S, which accounts for 6 percent of all G12 mutations. He and his colleagues have discovered that some chemical structures called beta lactones can specifically target serine residues in the active site of certain enzymes, including KRAS. By attaching beta lactones to a sotorasib-like backbone, Shokat and colleagues have developed a few potential compounds that they are currently fine-tuning.

“Mechanisms of resistance are the target of research that is ongoing and needs to be done so we can more effectively treat patients with KRAS mutations,” said Patricia LoRusso, DO, a professor of medicine and associate director of experimental therapeutics at Yale Cancer Center, who chaired the Anniversary Session. LoRusso also provided her insights into the future of KRAS targeting, particularly regarding combination therapy.
LoRusso praised the success of sotorasib as a monotherapy for non-small cell lung cancer given the long-term follow-up data reported at the meeting but explained that its durability may be limited by factors such as co-occurring mutations, the advanced tumor stage of patients offered sotorasib, and a lack of biomarkers of sustained response.
Currently, researchers are performing phase III clinical trials of sotorasib in combination with chemotherapy. LoRusso also mentioned combined inhibition of KRAS and other components of the KRAS pathway, including the cofactor SHP2, the downstream effector MEK, and the upstream activator EGFR. Immune checkpoint inhibitors, she explained, combined well with KRAS inhibitors in preclinical models, but the efficacy of the therapy is still being tested in clinical trials, such as the phase Ib/II trial CodeBreaK 101, which is examining a variety of drugs in combination with sotorasib.
“Will new KRAS-targeted agents and combinations enhance our therapeutic armamentarium against KRAS tumors? I think we have phenomenal scientists working in this arena that will help us say yes to that question,” LoRusso concluded.


