AACR COVID-19 and Cancer Virtual Meeting: Tracking the Evolution and Spread of SARS-CoV-2
In January, when most of us in the United States were busy living the hustle and bustle of everyday life, one shrewd virologist and computer biologist saw something that would quickly transform the way we function as a society. He started sounding the alarm to government agencies about an impending pandemic.
Trevor Bedford, PhD, associate professor in the Vaccine and Infectious Disease Division and the Computational Biology Program at Fred Hutchinson Cancer Research Center, is “one of the key opinion leaders about COVID-19,” said Program Committee Chair of the AACR Virtual Meeting: COVID-19 and Cancer and AACR President-Elect David Tuveson, MD, PhD, FAACR. Bedford is a Twitter influencer who uses the platform and his voice to disseminate factual, scientific information about the COVID-19 pandemic to his more than 259,000 followers. “He teaches the world through tweeting,” said Tuveson. Among his interesting approaches to studying the virus, Tuveson noted, months ago, Bedford correlated the spread of the virus to the use of the Open Table app and noticed increased incidence among people socializing at restaurants.
During the final day of the meeting, in a keynote address titled “Genomic Tracking of SARS-CoV-2 Evolution and Spread,” Bedford described the sophisticated computational and statistical methods he uses to map the phylogenetic tree of SARS-CoV-2 variants and track their spread to understand the underlying epidemic process.
Bedford began by explaining how the five genomes of novel SARS-CoV-2 that emerged in Wuhan, China, in January soon expanded to 12. The genomes lacked significant diversity, with no more than just a handful of mutations, suggesting that they had a recent common ancestor. His observations pointed to the fact that there was a single introduction of the virus into the human population during the November-December 2019 timeframe, capable of human-to-human epidemic spread.
“At this point, I freaked out,” Bedford said. He spent the week of January 20 alerting United States public health officials about what he found.
Bedford started updating and maintaining COVID-19 data on the nextstrain.org/ncov site, powered by the National Institutes of Health GenBank, an annotated collection of all publicly available DNA sequences, and GISAID, a global initiative for real-time data sharing of influenza virus and coronavirus genomic sequences. With Nextstrain, researchers conduct real-time molecular epidemiological and evolutionary analysis of the outbreak. Through this approach, Bedford and colleagues found that the infections that initially spread from Wuhan to Europe, Australia, and the United States in early January were caught and mostly stopped. However, other introductions of the virus to these parts of the world during the January-February timeframe became local epidemics in the months to come.
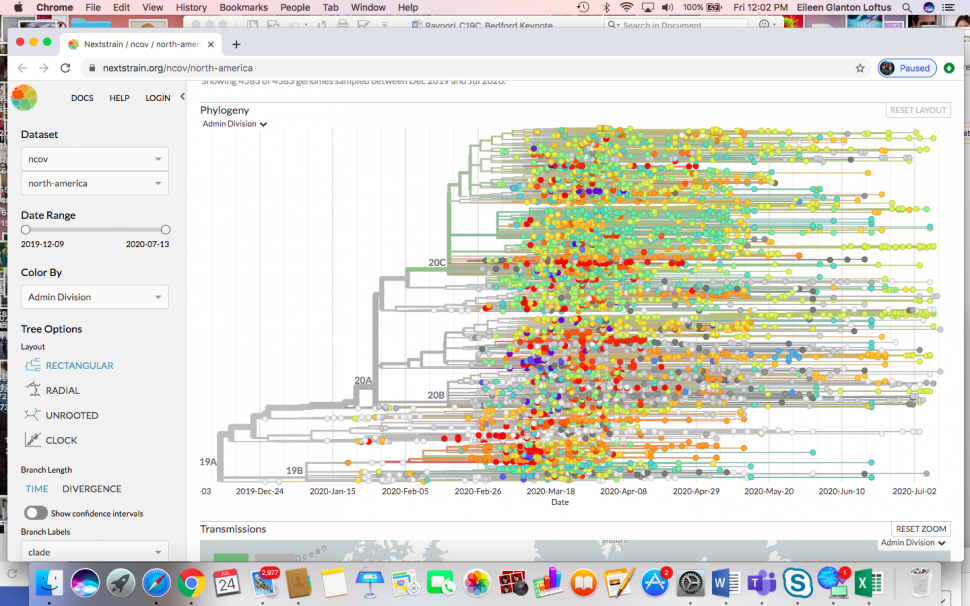
A snapshot of the virus spread in the United States in July shows that it is not just a single variant, but is, in fact, multiple variants and lineages that spread rapidly. These variants were localized and not imported, following travel restrictions.
Further studies on the functional aspects of the mutations revealed that a spike mutation, D614G, occurred early on in the epidemic and took root in Europe. The D614G variant is distinct from the ancestral variant that originated in Wuhan and was found to increase in frequency in the United States and Australia over time.
Data showed that with social distancing, the spread of the ancestral variant subsided more effectively than the D614G variant. The recent surges in Texas and other areas in the U.S. South are dominated by the D614G variant, Bedford noted in a discussion later. This is consistent with the knowledge that the D614G variant is more transmissible and yields a higher viral load than the ancestral variant. However, it is possible that this variant is prevalent because of multiple introductions in different regions of Europe, and subsequently to the United States, and not because of its functional aspects, according to Bedford.
“Moving forward, one thing that will be really useful is to be able to distinguish between endogenous spread and spread from importations,” Bedford said. Another important step is to integrate clinical data with mutation data to understand how the different variants impact clinical outcomes. This would also help establish other factors besides the functional aspects of the SARS-CoV-2 variants that could potentially influence outcomes, Bedford concluded. Bedford’s full presentation is available on Vimeo.