Editors’ Picks: February 2024 Highlights from AACR Journals
Roses are red, DAPI is blue, we have some Editors’ Picks for you …
Valentine’s Day may be over, but we’re still feeling the love for cancer research. February’s Editors’ Picks include insights into treatment resistance, clues about how cancer develops and spreads, and novel treatment approaches.
The abstracts of the selected studies are included below; for more details, follow the links to the full articles, which are freely available for a limited time.
Journal: Blood Cancer Discovery
Immunomodulatory drugs (IMiD) are a backbone therapy for multiple myeloma (MM). Despite their efficacy, most patients develop resistance, and the mechanisms are not fully defined. Here, we show that IMiD responses are directed by IMiD-dependent degradation of IKZF1 and IKZF3 that bind to enhancers necessary to sustain the expression of MYC and other myeloma oncogenes. IMiD treatment universally depleted chromatin-bound IKZF1, but eviction of P300 and BRD4 coactivators only occurred in IMiD-sensitive cells. IKZF1-bound enhancers overlapped other transcription factor binding motifs, including ETV4. Chromatin immunoprecipitation sequencing showed that ETV4 bound to the same enhancers as IKZF1, and ETV4 CRISPR/Cas9-mediated ablation resulted in sensitization of IMiD-resistant MM. ETV4 expression is associated with IMiD resistance in cell lines, poor prognosis in patients, and is upregulated at relapse. These data indicate that ETV4 alleviates IKZF1 and IKZF3 dependency in MM by maintaining oncogenic enhancer activity and identify transcriptional plasticity as a previously unrecognized mechanism of IMiD resistance.
Significance: We show that IKZF1-bound enhancers are critical for IMiD efficacy and that the factor ETV4 can bind the same enhancers and substitute for IKZF1 and mediate IMiD resistance by maintaining MYC and other oncogenes. These data implicate transcription factor redundancy as a previously unrecognized mode of IMiD resistance in MM.
This article was highlighted in the January issue. A commentary on the study can be found here. A related article was featured in last month’s edition of Editors’ Picks.
Journal: Cancer Discovery
Alveolar Differentiation Drives Resistance to KRAS Inhibition in Lung Adenocarcinoma
Lung adenocarcinoma (LUAD), commonly driven by KRAS mutations, is responsible for 7% of all cancer mortality. The first allele-specific KRAS inhibitors were recently approved in LUAD, but the clinical benefit is limited by intrinsic and acquired resistance. LUAD predominantly arises from alveolar type 2 (AT2) cells, which function as facultative alveolar stem cells by self-renewing and replacing alveolar type 1 (AT1) cells. Using genetically engineered mouse models, patient-derived xenografts, and patient samples, we found inhibition of KRAS promotes transition to a quiescent AT1-like cancer cell state in LUAD tumors. Similarly, suppressing Kras induced AT1 differentiation of wild-type AT2 cells upon lung injury. The AT1-like LUAD cells exhibited high growth and differentiation potential upon treatment cessation, whereas ablation of the AT1-like cells robustly improved treatment response to KRAS inhibitors. Our results uncover an unexpected role for KRAS in promoting intratumoral heterogeneity and suggest that targeting alveolar differentiation may augment KRAS-targeted therapies in LUAD.
Significance: Treatment resistance limits response to KRAS inhibitors in LUAD patients. We find LUAD residual disease following KRAS targeting is composed of AT1-like cancer cells with the capacity to reignite tumorigenesis. Targeting the AT1-like cells augments responses to KRAS inhibition, elucidating a therapeutic strategy to overcome resistance to KRAS-targeted therapy.
This article was highlighted in the February issue.
Journal: Cancer Epidemiology, Biomarkers & Prevention
Racial Disparities in Accessing Care along the Continuum of Cancer Genetic Service Delivery
Background: Public health calls to ensure equity in genomics and precision medicine necessitate a closer examination of how these efforts might differentially affect access to genetic services across demographic subgroups. This study set out to examine racial/ethnic disparities along the cancer genetic service delivery continuum.
Methods: Retrospective data are drawn from 15 clinical sites across 6 U.S. States. Individuals who screened at-risk for hereditary cancer were: (i) referred/scheduled to see a genetic counselor (referral workflow), or (ii) offered genetic testing at the point-of-care (POC testing workflow). Logistic regression analyses evaluated the associations between race/ethnicity and several outcomes including appointment scheduling, genetic counseling, and genetic testing, controlling for demographics, clinical factors, and county-level covariates.
Results: A total of 14,527 patients were identified at-risk. Genetic testing uptake was significantly higher at POC sites than referral sites (34% POC vs. 11% referral, P < 0.001). Race/ethnicity was significantly associated with testing uptake among all sites, with non-Hispanic Blacks having lower odds of testing compared with non-Hispanic Whites [aOR = 0.84; 95% confidence interval (CI), 0.71–1.00; P = 0.049]. Moreover, this disparity was observed at referral sites, but not POC sites. Among patients scheduled, non-Hispanic Blacks had lower odds of counseling (aOR = 0.28; 95% CI, 0.17–0.47; P < 0.001).
Conclusions: Findings suggest that factors influencing genetic counseling show rates may be driving disparities in genetic testing.
Impact: Strategies to reduce barriers to seeing a genetic counselor, including modifications to clinical workflow, may help mitigate racial/ethnic disparities in genetic testing.
This article was highlighted in the January issue.
Journal: Cancer Immunology Research
RALDH1 Inhibition Shows Immunotherapeutic Efficacy in Hepatocellular Carcinoma
Globally, hepatocellular carcinoma (HCC) is one of the most commonly diagnosed cancers and a leading cause of cancer-related death. We previously identified an immune evasion pathway whereby tumor cells produce retinoic acid (RA) to promote differentiation of intratumoral monocytes into protumor macrophages. Retinaldehyde dehydrogenase 1 (RALDH1), RALDH2, and RALDH3 are the three isozymes that catalyze RA biosynthesis. In this study, we have identified RALDH1 as the key driver of RA production in HCC and demonstrated the efficacy of RALDH1-selective inhibitors (Raldh1-INH) in suppressing RA production by HCC cells. Raldh1-INH restrained tumor growth in multiple mouse models of HCC by reducing the number and tumor-supporting functions of intratumoral macrophages as well as increasing T-cell infiltration and activation within tumors. Raldh1-INH also displayed favorable pharmacokinetic, pharmacodynamic, and toxicity profiles in mice thereby establishing them as promising new drug candidates for HCC immunotherapy.
Journal: Cancer Prevention Research
Risk and outcome of acute promyelocytic leukemia (APL) are particularly worsened in obese-overweight individuals, but the underlying molecular mechanism is unknown. In established mouse APL models (Ctsg-PML::RARA), we confirmed that obesity induced by high-fat diet (HFD) enhances leukemogenesis by increasing penetrance and shortening latency, providing an ideal model to investigate obesity-induced molecular events in the preleukemic phase. Surprisingly, despite increasing DNA damage in hematopoietic stem cells (HSC), HFD only minimally increased mutational load, with no relevant impact on known cancer-driving genes. HFD expanded and enhanced self-renewal of hematopoietic progenitor cells (HPC), with concomitant reduction in long-term HSCs. Importantly, linoleic acid, abundant in HFD, fully recapitulates the effect of HFD on the self-renewal of PML::RARA HPCs through activation of peroxisome proliferator-activated receptor delta, a central regulator of fatty acid metabolism. Our findings inform dietary/pharmacologic interventions to counteract obesity-associated cancers and suggest that nongenetic factors play a key role.
Prevention Relevance: Our work informs interventions aimed at counteracting the cancer-promoting effect of obesity. On the basis of our study, individuals with a history of chronic obesity may still significantly reduce their risk by switching to a healthier lifestyle, a concept supported by evidence in solid tumors but not yet in hematologic malignancies.
This study was featured on the cover of the February issue. A related commentary can be found here.
Journal: Cancer Research (February 1 issue)
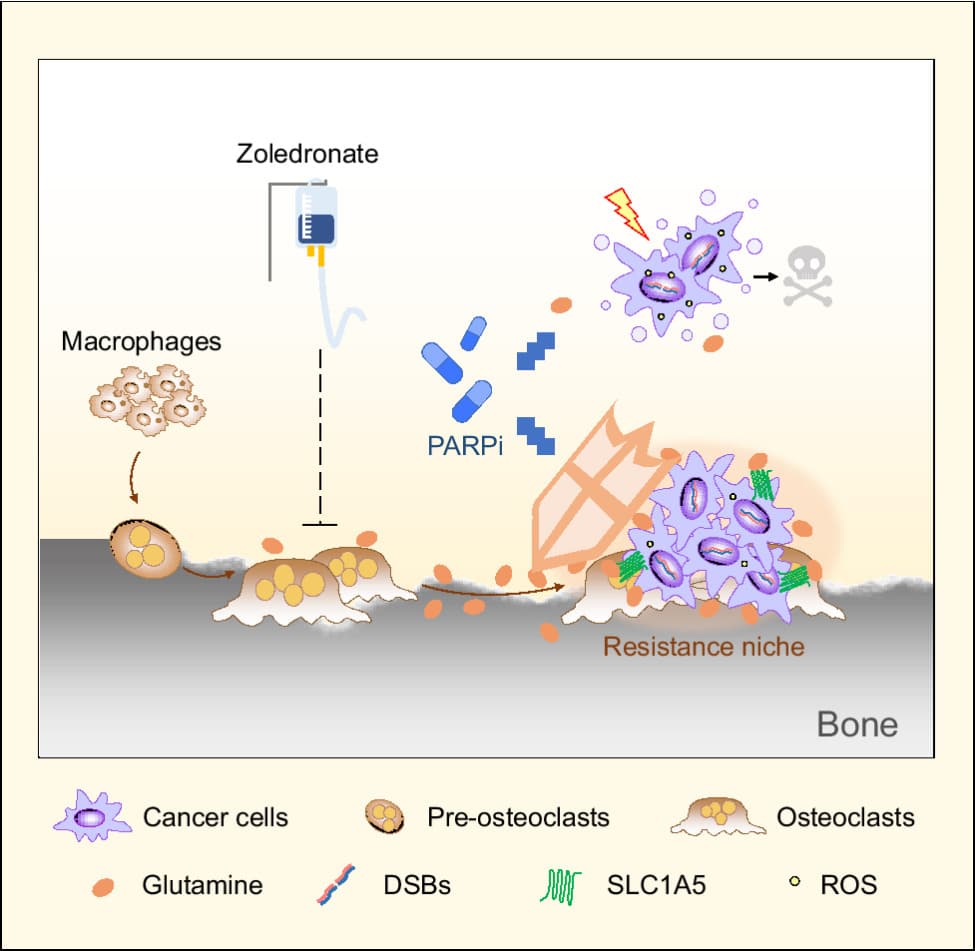
The majority of patients with late-stage breast cancer develop distal bone metastases. The bone microenvironment can affect response to therapy, and uncovering the underlying mechanisms could help identify improved strategies for treating bone metastatic breast cancer. Here, we observed that osteoclasts reduced the sensitivity of breast cancer cells to DNA damaging agents, including cisplatin and the PARP inhibitor (PARPi) olaparib. Metabolic profiling identified elevated glutamine production by osteoclasts. Glutamine supplementation enhanced the survival of breast cancer cells treated with DNA damaging agents, while blocking glutamine uptake increased sensitivity and suppressed bone metastasis. GPX4, the critical enzyme responsible for glutathione oxidation, was upregulated in cancer cells following PARPi treatment through stress-induced ATF4-dependent transcriptional programming. Increased glutamine uptake and GPX4 upregulation concertedly enhanced glutathione metabolism in cancer cells to help neutralize oxidative stress and generate PARPi resistance. Analysis of paired patient samples of primary breast tumors and bone metastases revealed significant induction of GPX4 in bone metastases. Combination therapy utilizing PARPi and zoledronate, which blocks osteoclast activity and thereby reduces the microenvironmental glutamine supply, generated a synergistic effect in reducing bone metastasis. These results identify a role for glutamine production by bone-resident cells in supporting metastatic cancer cells to overcome oxidative stress and develop resistance to DNA-damaging therapies.
Significance: Metabolic interaction between osteoclasts and tumor cells contributes to resistance to DNA-damaging agents, which can be blocked by combination treatment with PARP and osteoclast inhibitors to reduce bone metastatic burden.
Journal: Cancer Research (February 15 issue)
Mesothelin Secretion by Pancreatic Cancer Cells Co-opts Macrophages and Promotes Metastasis
Pancreatic ductal adenocarcinoma (PDAC) is a highly metastatic disease, yet effective treatments to inhibit PDAC metastasis are lacking. The rich PDAC tumor microenvironment plays a major role in disease progression. Macrophages are the most abundant immune cell population in PDAC tumors and can acquire a range of functions that either hinder or promote tumor growth and metastasis. Here, we identified that mesothelin secretion by pancreatic cancer cells co-opts macrophages to support tumor growth and metastasis of cancer cells to the lungs, liver, and lymph nodes. Mechanistically, secretion of high levels of mesothelin by metastatic cancer cells induced the expression of VEGF alpha (VEGFA) and S100A9 in macrophages. Macrophage-derived VEGFA fed back to cancer cells to support tumor growth, and S100A9 increased neutrophil lung infiltration and formation of neutrophil extracellular traps. These results reveal a role for mesothelin in regulating macrophage functions and interaction with neutrophils to support PDAC metastasis.
Significance: Mesothelin secretion by cancer cells supports pancreatic cancer metastasis by inducing macrophage secretion of VEGFA and S100A9 to support cancer cell proliferation and survival, recruit neutrophils, and stimulate neutrophil extracellular trap formation.
A related commentary can be found here.
Journal: Clinical Cancer Research (February 1 issue)
Purpose: Mutations in BTK, PLCG2, and BCL2 have been reported in patients with progressive disease (PD) on continuous single-agent BTK or BCL2 inhibitor treatment. We tested for these mutations in samples from patients with PD after completion of first-line treatment with fixed-duration ibrutinib plus venetoclax for chronic lymphocytic leukemia (CLL) in the phase II CAPTIVATE study.
Patients and Methods: A total of 191 patients completed fixed-duration ibrutinib plus venetoclax (three cycles of ibrutinib then 12–13 cycles of ibrutinib plus venetoclax). Genomic risk features [del(11q), del(13q), del(17p), trisomy 12, complex karyotype, unmutated IGHV, TP53 mutated] and mutations in genes recurrently mutated in CLL (ATM, BIRC3, BRAF, CHD2, EZH2, FBXW7, MYD88, NOTCH1, POT1, RPS15, SF3B1, XPO1) were assessed at baseline in patients with and without PD at data cutoff; gene variants and resistance-associated mutations in BTK, PLCG2, or BCL2 were evaluated at PD.
Results: Of 191 patients completing fixed-duration ibrutinib plus venetoclax, with median follow-up of 38.9 months, 29 (15%) developed PD. No baseline risk feature or gene mutation was significantly associated with development of PD. No previously reported resistance-associated mutations in BTK, PLCG2, or BCL2 were detected at PD in 25 patients with available samples. Of the 29 patients with PD, 19 have required retreatment (single-agent ibrutinib, n = 16, or ibrutinib plus venetoclax, n = 3); 17 achieved partial response or better, 1 achieved stable disease, and 1 is pending response assessment.
Conclusions: First-line fixed-duration combination treatment with ibrutinib plus venetoclax may mitigate development of resistance mechanisms associated with continuous single-agent targeted therapies, allowing for effective retreatment.
This study was highlighted in the February 1 issue. A related commentary can be found here.
Journal: Clinical Cancer Research (February 15 issue)
Purpose: We conducted research on CDK4/6 inhibitors (CDK4/6i) simultaneously in the preclinical and clinical spaces to gain a deeper understanding of how senescence influences tumor growth in humans.
Patients and Methods: We coordinated a first-in-kind phase II clinical trial of the CDK4/6i abemaciclib for patients with progressive dedifferentiated liposarcoma (DDLS) with cellular studies interrogating the molecular basis of geroconversion.
Results: Thirty patients with progressing DDLS enrolled and were treated with 200 mg of abemaciclib twice daily. The median progression-free survival was 33 weeks at the time of the data lock, with 23 of 30 progression-free at 12 weeks (76.7%, two-sided 95% CI, 57.7%–90.1%). No new safety signals were identified. Concurrent preclinical work in liposarcoma cell lines identified ANGPTL4 as a necessary late regulator of geroconversion, the pathway from reversible cell-cycle exit to a stably arrested inflammation-provoking senescent cell. Using this insight, we were able to identify patients in which abemaciclib induced tumor cell senescence. Senescence correlated with increased leukocyte infiltration, primarily CD4-positive cells, within a month of therapy. However, those individuals with both senescence and increased TILs were also more likely to acquire resistance later in therapy. These suggest that combining senolytics with abemaciclib in a subset of patients may improve the duration of response.
Conclusions: Abemaciclib was well tolerated and showed promising activity in DDLS. The discovery of ANGPTL4 as a late regulator of geroconversion helped to define how CDK4/6i-induced cellular senescence modulates the immune tumor microenvironment and contributes to both positive and negative clinical outcomes.
This article was highlighted in the February 15 issue. A related commentary can be found here.
Journal: Molecular Cancer Research
Breast cancer is the most common cancer in females, affecting one in every eight women and accounting for the majority of cancer-related deaths in women worldwide. Germline mutations in the BRCA1 and BRCA2 genes are significant risk factors for specific subtypes of breast cancer. BRCA1 mutations are associated with basal-like breast cancers, whereas BRCA2 mutations are associated with luminal-like disease. Defects in mammary epithelial cell differentiation have been previously recognized in germline BRCA1/2 mutation carriers even before cancer incidence. However, the underlying mechanism is largely unknown. Here, we employ spatial transcriptomics to investigate defects in mammary epithelial cell differentiation accompanied by distinct microenvironmental alterations in preneoplastic breast tissues from BRCA1/2 mutation carriers and normal breast tissues from noncarrier controls. We uncovered spatially defined receptor–ligand interactions in these tissues for the investigation of autocrine and paracrine signaling. We discovered that β1-integrin-mediated autocrine signaling in BRCA2-deficient mammary epithelial cells may differ from BRCA1-deficient mammary epithelial cells. In addition, we found that the epithelial-to-stromal paracrine signaling in the breast tissues of BRCA1/2 mutation carriers is greater than in control tissues. More integrin–ligand pairs were differentially correlated in BRCA1/2-mutant breast tissues than noncarrier breast tissues with more integrin receptor-expressing stromal cells.
Implications: These results suggest alterations in the communication between mammary epithelial cells and the microenvironment in BRCA1 and BRCA2 mutation carriers, laying the foundation for designing innovative breast cancer chemo-prevention strategies for high-risk patients.
This article was featured on the cover and highlighted in the February issue.
Journal: Molecular Cancer Therapeutics
CD38 x ICAM-1 Bispecific Antibody Is a Novel Approach for Treating Multiple Myeloma and Lymphoma
The cluster of differentiation 38 (CD38) is a well-validated target for treating multiple myeloma. Although anti-CD38 mAbs have demonstrated outstanding initial responses in patients with multiple myeloma, nearly all patients eventually develop resistance and relapse. In addition, currently approved CD38 targeting therapies have failed to show monotherapy efficacy in lymphomas, where CD38 expression is present but at lower levels. To effectively target CD38 on tumor cells, we generated an antibody-dependent cellular cytotoxicity (ADCC) enhanced bispecific CD38 x intercellular cell adhesion molecule 1 (ICAM-1) antibody, VP301. This bispecific antibody targets unique epitopes on CD38 and ICAM-1 on tumor cells with reduced red blood cell binding compared with the benchmark CD38 antibody daratumumab. VP301 demonstrated potent ADCC and antibody-dependent cellular phagocytosis activities on a selected set of myeloma and lymphoma cell lines even those with low CD38 expression. In an ex vivo drug sensitivity assay, we observed responses to VP301 in multiple myeloma primary samples from relapsed/refractory patients. Moreover, VP301 demonstrated potent tumor inhibition activities in in vivo myeloma and lymphoma models. Interestingly, combination of VP301 with the immunomodulatory drug, lenalidomide, led to synergistic antitumor growth activity in an in vivo efficacy study. In conclusion, the CD38 x ICAM-1 bispecific antibody VP301 demonstrated promising efficacy and specificity toward CD38+ and ICAM-1+ tumor cells and represents a novel approach for treating multiple myeloma and lymphoma.
This article was featured on the cover and highlighted in the February issue.
Journal: Cancer Research Communications
Neuroblastoma is the most common extracranial tumor, accounting for 15% of all childhood cancer-related deaths. The long-term survival of patients with high-risk tumors is less than 40%, and MYCN amplification is one of the most common indicators of poor outcomes. Zika virus (ZIKV) is a mosquito-borne flavivirus associated with mild constitutional symptoms outside the fetal period. Our published data showed that high-risk and recurrent neuroblastoma cells are permissive to ZIKV infection, resulting in cell type–specific lysis. In this study, we assessed the efficacy of ZIKV as an oncolytic treatment for high-risk neuroblastoma using in vivo tumor models. Utilizing both MYCN-amplified and non-amplified models, we demonstrated that the application of ZIKV had a rapid tumoricidal effect. This led to a nearly total loss of the tumor mass without evidence of recurrence, offering a robust survival advantage to the host. Detection of the viral NS1 protein within the tumors confirmed that a permissive infection preceded tissue necrosis. Despite robust titers within the tumor, viral shedding to the host was poor and diminished rapidly, correlating with no detectable side effects to the murine host. Assessments from both primary pretreatment and recurrent posttreatment isolates confirmed that permissive sensitivity to ZIKV killing was dependent on the expression of CD24, which was highly expressed in neuroblastomas and conferred a proliferative advantage to tumor growth. Exploiting this viral sensitivity to CD24 offers the possibility of its use as a prognostic target for a broad population of expressing cancers, many of which have shown resistance to current clinical therapies.
Significance: Sensitivity to the tumoricidal effect of ZIKV on high-risk neuroblastoma tumors is dependent on CD24 expression, offering a prognostic marker for this oncolytic therapy in an extensive array of CD24-expressing cancers.