Dragging Oncogenes From the Cell: A New Horizon for Targeting KRAS
Thousands of years ago, the ancient Greeks waged a decadelong siege against the seemingly impenetrable city of Troy.
The massive stone walls and carefully guarded gates made it impossible for the Greeks to enter the city and defeat their enemies. In order to finally end the war, they disguised themselves as something authorized to enter the city—their famed Trojan horse.
To cancer researchers, the cell membrane functions somewhat like Troy’s walls, barring some potential therapies from entry. Antibodies, for example, can be designed to neutralize virtually any protein, but they are too large to pass through the cell membrane without specialized help.
If only researchers could deliver an antibody that was authorized to enter the cell.
Jose Conejo-Garcia, MD, PhD, a professor of immunology at the Duke University School of Medicine and a member of the Duke Cancer Institute, may have found a way to make this Trojan horse dream a reality. He and his colleagues have discovered that an antibody known as IgA can be brought into the cells that comprise many types of solid tumors. Essentially, IgA antibodies can be designed to target specific cancer driver proteins and sent in as Trojan horses.
But unlike the Greeks, these IgA antibodies don’t destroy the cancer cells; they latch onto their target proteins and drag them out of the cell to dampen tumor growth. It would be as if the Greeks had grabbed the Trojan generals, pulled them into the horse, and wheeled them out the other side of the city.
Conejo-Garcia presented these findings at the AACR Annual Meeting 2023 and the 2023 Special Conference in Endometrial Cancer, and the data were recently published in the journal Immunity. In an era focused on mitigating the activity of elusive “undruggable” proteins such as KRAS, could these specialized Trojan horses present a new therapeutic opportunity?
What Is IgA?
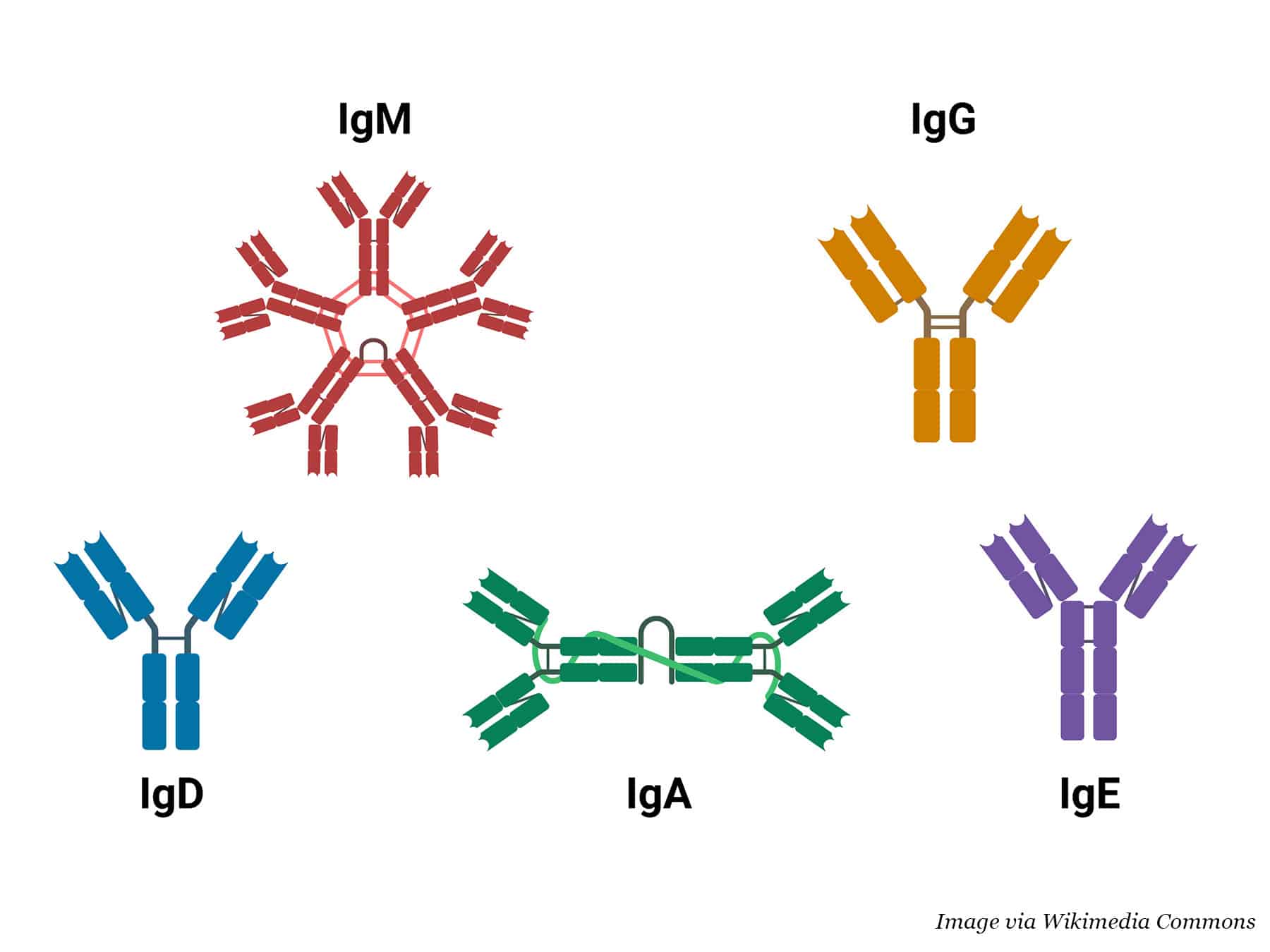
Antibodies are part of the body’s natural defense against infections, foreign substances, and cancer. They identify and bind to potential pathogens, tagging them for destruction by other immune cells. The human immune system uses five main types of antibodies: IgA, IgG, IgD, IgE, and IgM.
Researchers have been using therapeutic antibodies—antibodies that are designed and manufactured to target cancer-specific proteins—to treat cancer for nearly 30 years. These engineered antibodies generally bind to receptors on the cell membrane that drive cancer growth. They not only stimulate antitumor immune responses but can also block the activity of these receptors. All therapeutic antibodies currently approved by the U.S. Food and Drug Administration (FDA) are of the IgG subclass due to its optimal properties for activating downstream immune pathways.
One challenge with IgG antibodies: They are too large to pass through the cell membrane and can therefore only target cancer drivers on the outside of the cell, while many cancer drivers exist inside the cell. Targeting intracellular proteins has thus far relied on small molecule inhibitors, but they are not as versatile as antibodies. Whereas antibodies can be engineered to target nearly any part of a protein’s surface, small molecules must fit in an available binding pocket that disrupts either the protein’s activation or downstream activity. Several cancer drivers are currently considered “undruggable” for their lack of drug-binding pockets.
An antibody that can travel inside the cell and neutralize driver proteins, therefore, might provide an ideal solution.
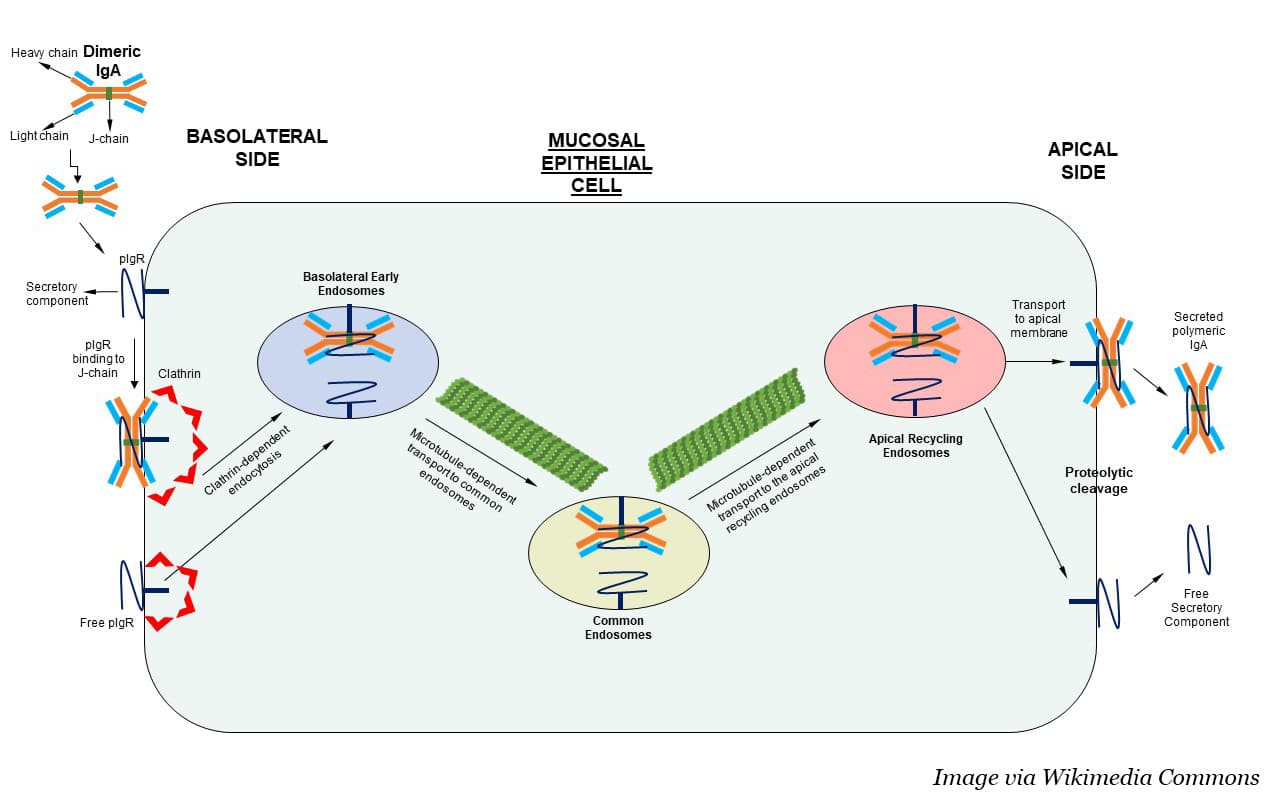
Near epithelial cells, IgA exists in a dimerized form, with two molecules held together by a J chain. Dimeric IgA (dIgA) can bind to the polymeric immunoglobulin receptor (PIGR) and enter the cell through a process called receptor-mediated endocytosis. IgA-bound PIGR essentially stimulates the surrounding areas of the cell membrane to envelop the receptor and pull it into the cell, where it passes through the cell’s interior in a series of membrane-bound bubbles. PIGR and dimeric IgA (dIgA) then exit the cell on the other side.
Can this process be harnessed to neutralize intracellular cancer driver proteins? Conejo-Garcia and colleagues asked. On its way through the cytoplasm, could the internalized dIgA latch onto a target protein and pull it out of the cell?
A KRAS Grab-and-go
Conejo-Garcia and colleagues set their sights on a notoriously difficult protein to neutralize, the nearly ubiquitous cancer driver KRAS. Mutated in around 20% of solid tumors, KRAS was believed to have an “undruggable” protein surface until the first targeted therapies, sotorasib (Lumakras) and adagrasib (Krazati) were approved by the FDA in 2021 and 2022, respectively.
Sotorasib and adagrasib target the same KRAS mutation, called G12C, which makes up approximately 12% of KRAS mutations across all cancer types and is especially common in lung cancer. However, two more common mutations—G12D and G12V (33% and 23% of all KRAS mutations, respectively)—do not have any FDA-approved targeted therapies. Neither do the various other mutations that can occur in the KRAS gene.
Further, only about one-third of patients with tumors approved for sotorasib treatment respond to the drug, and many who do respond eventually develop resistance.

Conejo-Garcia and colleagues designed a dIgA antibody that binds to KRASG12D but not to unmutated KRAS, which is found in healthy tissues. Using a cell model of ovarian cancer, the researchers showed that treatment with targeted dIgA decreased the amount of KRASG12D inside the cells and increased the amount outside the cells. When the cells were plated across an impenetrable membrane to divide a well into two chambers, and KRASG12D-targeted dIgA was added to the top chamber, KRASG12D was later found in the bottom chamber, confirming that the migration of dIgA through the cell is directional.
When ovarian cancer cells were xenografted into mice, both intratumoral and intraperitoneal injection of KRASG12D-targeted dIgA decreased tumor growth, while IgG and untargeted dIgA did not. Using data from The Cancer Genome Atlas, Conejo-Garcia and colleagues found that PIGR is highly expressed in virtually all epithelial malignancies (and B-cell lymphoma), indicating that this technique may work in tumor types beyond ovarian cancer.
The researchers tested their targeted dIgA in mouse models of endogenous KRASG12D-driven lung cancer and saw similar tumor shrinkage. In mouse models with intact immune systems, the existence of CD8+ T cells enhanced the antitumor effects of the targeted dIgA, suggesting that the engineered dIgA molecules can stimulate immune responses in addition to neutralizing mutant KRAS.
In both ovarian and lung cancer models, a small-molecule inhibitor of KRASG12D performed similarly to the dIgA, but 20 daily injections were required to produce the same effect as five injections of the targeted dIgA. Conejo-Garcia and colleagues emphasized that the half-life of dIgA (around six days in primates) is significantly longer than most small molecule inhibitors (around six hours), which may help account for the sustained effects.
“We honestly believe that this can change the way that we treat cancer and pave the way for a new generation of immunotherapies,” Conejo-Garcia said during his Annual Meeting presentation.
Unanswered Questions
One aspect of the mechanism underlying the efficacy of KRASG12D-targeted dIgA particularly surprised Conejo-Garcia and colleagues. When dIgA migrates through the cell, it does so enveloped in a cargo vesicle, which should effectively separate dIgA from other cellular contents. However, when the researchers tagged KRASG12D and their dIgA molecules with fluorescent dyes, they found that the two colocalized within the vesicles.
How does dIgA pull mutant KRAS into the vesicles? Conejo-Garcia and colleagues hypothesized that because KRAS exists close to the cell membrane, it might sometimes get swept into the vesicles alongside the dIgA molecules. Alternatively, other vesicles transporting KRAS back to the cell surface may merge with the ones holding dIgA.
In a follow-up experiment, however, the researchers designed a dIgA specific to mutant IDH1, a protein that exists deep in the cytoplasm. In mouse cell xenograft models of colorectal cancer, treatment with IDH1-targeted dIgA decreased tumor size specifically in tumors harboring IDH1 mutations. How dIgA was able to colocalize with a protein that goes nowhere near the cell membrane remains a mystery, but Conejo-Garcia and colleagues intend to follow up on this question in future experiments.
It also remains to be seen whether tumors will develop resistance to targeted dIgA by downregulating PIGR expression, the researchers noted. They suggested that combining dIgA treatment with small molecule inhibitors may help delay or prevent resistance by targeting the cancer driver through different simultaneous mechanisms. The researchers also stated that the safety of such a therapy would need to be evaluated in humans, but they cited some promising data. Antibody preparations containing IgA and IgG mixtures have been used to treat certain infectious diseases with limited toxicity.
Plenty more research is needed, but in the continuing quest to confront seemingly indomitable cancer drivers, perhaps a sneaky approach from ancient Greece may provide a promising path forward.